Table of Contents
Page created on October 6, 2018. Last updated on December 18, 2024 at 16:56
Because the adrenal gland produces mineralocorticoids like aldosterone is it also often involved in secondary hypertension. Conn syndrome, Cushing syndrome, adrenogenital syndrome, pheochromocytoma, acromegaly and thyroid problems all cause secondary hypertension. Let’s see how.
Conn syndrome
Conn syndrome, or primary hyperaldosteronism, increases blood pressure by increased levels of mineralocorticoids. These hormones, especially aldosterone, cause increased salt and water retention in the kidneys. This causes plasma volume to initially become increased, which also increases the blood pressure.
The increased plasma volume causes increased venous return and therefore increased stroke volume and cardiac output. The increased cardiac output causes all tissues to become hyperperfused, which they don’t want. Organs will react by arteriolar vasoconstriction, which will cause perfusion to return to normal, but an increased total peripheral resistance. The cardiac output will therefore normalize, but the increased resistance still causes hypertension.
A mechanism called the aldosterone escape mechanism prevents the fluid retention in Conn syndrome from being excessive. The incresed plasma volume will stimulate production of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). These peptides will “counteract” the salt-retaining effect of aldosterone by inducing diuresis. Potassium and protons will still be lost by the kidneys.
Cushing syndrome
Recall that cortisol can, to some extent, bind to mineralocorticoid receptors. If the level of cortisol is high, like in Cushing disease or Cushing syndrome will same effects as in Conn syndrome be observed.
Cortisol also causes increased sensitivity to catecholamines, which enhances their hypertensive effect. It also increases angiotensinogen production by the liver. Because of these effects (and the lack of aldosterone escape mechanism here) is the blood pressure in Cushing’s syndrome higher than in Conn syndrome.
Adrenogenital syndrome
In adrenogenital syndrome, or congenital adrenal hyperplasia, will the adrenal glands grow due to hyperplasia. This can occur due to the deficiency of either 21-hydroxylase, 11β-hydroxylase or 17α-hydroxylase. However, hypertension is only seen in deficiency of 11β-hydroxylase or 17α-hydroxylase.
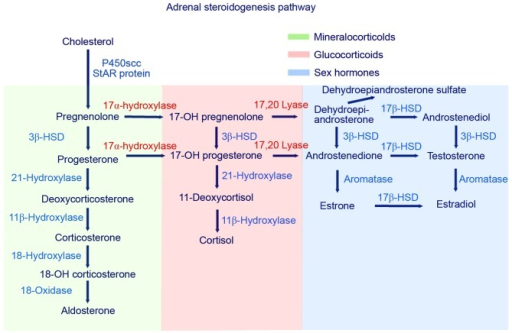
As can be seen on the figure, if any of these enzymes are deficient will the production of cortisol be deficient as well. When cortisol is deficient will ACTH levels increase because of a lack of negative feedback. It is this increase in ACTH that causes the adrenal glands to hyperplasia.
In deficiency of 11β-hydroxylase or 17α-hydroxylase the level of deoxycorticosterone (DOC) will be very high. DOC is a weak mineralocorticoid which has no real significance in physiological state, but when its level is very high it becomes significant enough to cause hypertension by activating mineralocorticoid receptors.
Apparent mineralocortioid excess syndrome
AME syndrome is a condition where the hormone 11β-hydroxysteroid dehydrogenase is deficient. This hormone usually inactivates cortisol in the kidneys, so that cortisol doesn’t bind too much to mineralocorticoid receptors. When this enzyme is deficient will cortisol have an increased effect on mineralocorticoid receptors in the kidney, making it have a similar effect as aldosterone.
Pheochromocytoma
A pheochromocytoma is a catecholamine-secreting tumour typically found in the adrenal medulla. They are usually benign.
In healthy patients is the ratio between noradrenaline and adrenaline in the blood 1:5, meaning that there is five times as much adrenaline than noradrenaline. However, in the presence of a pheochromocytoma is this ratio shifted to 1:1, meaning that the amount of noradrenaline and adrenaline in the blood is now the same. Noradrenaline has a higher effect on α1-adrenergic receptors than adrenaline does, which is the receptor that causes vasoconstriction.
In most cases the blood pressure is usually normal, but the patient experiences episodes of sudden, explosive (paroxysmal) increase in blood pressure, even up to 300/150 mmHg!
Diagnosis is made by the detection of noradrenaline and adrenaline breakdown products in a 24 hour urine sample. Treatment is α-blockers and removal of the tumour.
It is important to note that 21β-hydroxysteroid dehydrogenase deficiency also causes hypertension by reduced neutralization of cortisol effect on mineralocorticoid receptors in the kidneys.
Added, and fixed the spelling error from the book (21 to 11).