Table of Contents
Page created on January 11, 2021. Last updated on June 7, 2023 at 13:49
NEONATOLOGY
1. Classification of newborns. Basic concepts of perbrinatology
- Important terms
- Live birth = presence of vital signs at birth
- Miscarriage = absence of vital signs and pregnancy loss before week 20
- Stillbirth = absence of vital signs and pregnancy loss after week 20
- Classification according to age
- Newborn = child < 28 days of age
- Infant = child < 1 year of age
- Classification according to term
- Preterm infant = infant born before 37 weeks of gestation
- Term infant = infant born between 37 and 42 weeks of gestation
- Post term infant = infant born after 42 weeks of gestation
- Classification according to weight depending on gestational age
- Small-for-gestational age (SGA) infant = birthweight < 10th percentile for gestational age
- Appropriate-for-gestational age (AGA) infant = birthweight 10th – 90th percentile
- Large-for-gestational age (LGA) infant = birthweight > 90th percentile
- Classification according to weight independent of gestational age
- Low birth weight = birthweight 1500 – 2500 g
- Very low birth weight = birthweight 1000 – 1500 g
- Extremely low birth weight = birthweight < 1000 g
- Definitions
- Congenital = contracted in utero
- Perinatal = from 24 weeks of gestation until 4 weeks after birth
- Antenatal = before birth
- Postnatal = after birth
- Neonatal = from birth until 4 weeks after birth
2. The characteristics of the premature and intrauterine growth restriction infants
Characteristics of premature infants
- Premature (pre-term) infant
- Physical characteristics
- Big head, short trunk
- Immature extremities
- Nails not covering tip of fingers
- Oedematous, pinkish, thin skin with subcutaneous fat missing
- No testes in scrotum
- Presence of lanugo (small hairs)
- Labia majora don’t cover labia minora
- Few palmar creases
- Immature breathing mechanism (weak cry) -> require artificial respiration
- Immature heat regulation -> require incubator for weeks or months
- Immature GI tract -> require parenteral nutrition
- Physical characteristics
Intrauterine growth restriction
Intrauterine growth restriction (IUGR) or foetal growth restriction (FGR) is a pathological state where the foetus does not achieve its intrauterine growth potential. In other words, the foetus does not grow as much as they would if all factors were optimal. In most cases, IUGR causes a foetus which is small for gestational age (SGA). Small for gestational age is defined as an estimated foetal weight which is less than the 10th percentile for that gestational age and gender.
However, not IUGR does not always cause SGA. A foetus which was “destined” to be larger than the average (comes from a large family) but for some reason had their intrauterine growth restricted, may have a weight as appropriate for gestational age (AGA). Likewise, some foetuses are small for gestational age but without IUGR, likely because they come from a family of smaller people. These are called constitutionally small for gestational age.
To summarise, IUGR is always pathological, while SGA isn’t necessarily. It’s estimated that 25 – 50% of SGA foetuses are simply constitutionally small. Also, IUGR doesn’t always cause SGA, but in most cases it does. IUGR newborns are sometimes called dysmature.
Etiology
- Foetal
- TORCH infection
- Chromosomal abnormality
- Multiple gestation
- Placental
- Abnormal placenta or placentation
- Maternal
- Previous child with IUGR
- Mother was growth restricted herself
- Preeclampsia
- Maternal chronic disease
- Exposure to environmental factors (smoking, tobacco, pollution, drugs)
Classification
We distinguish symmetrical and asymmetrical IUGR.
Symmetric IUGR (or early IUGR) refers to IUGR where the whole body is smaller, but proportional. This accounts for only 20 – 30% of cases and is usually due to foetal causes. It typically occurs in the early stages of gestation.
Asymmetrical IUGR (or late IUGR) refers to IUGR where the body is disproportionally smaller than the head. This accounts for most cases of IUGR and is usually due to placental or maternal causes, and typically occurs in the later stages of gestation.
Characteristics of dysmature newborns
Dysmature newborns are often small for gestational age (SGA). In case of asymmetrical IUGR the dimensions of the head are normal while the body and limbs are small. In case of symmetrical IUGR the entire body is proportionally small. These newborns may also appear thin and malnourished.
Prognosis
The presence of IUGR indicates an increased risk for adverse outcomes, including abnormal neurodevelopment and death. There may also be longer term outcomes, including obesity, diabetes, cardiovascular disease, etc.
3. The characteristics of the mature newborn
- Measurements
- Bodyweight – 3000 – 4000 g
- Length – 47 – 55 cm
- Head circumference – ~36 cm
- Heart rate – 120-160/min
- Respiratory rate – 40-60/min
- Physical characteristic
- Nails cover tip of fingers
- Good skin turgor
- Body covered by mucous
- Testicles in scrotum
- Labia minora covered by labia majora
- Strong cry immediately after birth
- Plantar creases cover the soles of the feet
- Reacts to strong stimuli
- React to the environment (e.g. taste awareness)
4. The first evaluation of the newborn baby, routine delivery room and initial care
- See also topic B12 from ob/gyn 1 which explains it better.
- First evaluation
- Ask the following three questions.
- Is the infant term?
- Does the infant have good muscle tone?
- Is the infant breathing or crying?
- If answers to all are “yes”, the newborn does not need resuscitation. Proceed to routine care
- If not, stabilize as necessary
- Maintain temperature
- A – clear airways
- B – breathing – ventilation and oxygenation
- C – chest compressions
- Administer epinephrine or fluids
- Ask the following three questions.
- Routine care
- Drying the newborn
- Clearing airway secretions
- By wiping, not by suction
- Maintain temperature
- Clamp and cut the umbilical cord
- Skin-to-skin contact with mother
- Vitamin K shot
- Ophthalmic antibiotic drops
- Apgar score
- Performed at 1 and 5 minutes after birth
- Used to evaluate status of the newborn and its adaptation to the environment
- Score
- Normal: 7 -10
- Moderately abnormal: 4 – 6
- Low – 0 – 3
| Component | 0 points | 1 point | 2 points |
| Appearance (skin colour) | Cyanotic or pale | Acrocyanosis | Pink body and extremities |
| Pulse (heart rate) | None | < 100/min | > 100/min |
| Grimace (reflex response to irritable stimuli) | None | Whimpering, grimace | Crying, active withdrawal |
| Activity (muscle tone, movement) | No movement, hypotonia | Some flexion | Active motion |
| Respirations | None | Weak crying, irregular/slow/weak breathing or gasping | Regular breathing, strong cry |
- Other
- General appearance
- Is the newborn well, cooperative, agitated, weak, irritable, serious, life-threatening, etc.
- Sex
- Identification of deformations or malformations
- Determination of state of foetal nutrition
- Amount of subcutaneous fat on thigh and glutes
- Amount of Wharton’s jelly in umbilical cord
- Signs of respiratory distress
- Paradoxical breathing is normal
- Measurements
- Length
- Weight
- Head circumference
- Vital signs
- Axillary temp (36,5 – 37,5)
- Respiratory rate (40 – 60)
- Heart rate (120 – 160)
- Blood pressure (65-85/45-55)
- Skin
- Colour
- Should be rose-pale
- Acrocyanosis normal in the first days
- Central cyanosis never normal
- Jaundice in the first 24 hours is pathological
- Physiological jaundice in days 3 – 8
- Greenish discoloration is meconium
- Lesions or pigmentation changes
- Examine turgor
- Colour
- Head
- Size and shape
- Palpate fontanelles
- Bulging when infant is sitting -> Increased ICP
- Depressed -> dehydration
- Palpate sutures
- Face and mouth
- Facial palsy
- Position, symmetry of eyes
- Examination of the red-light reflex
- Colour of lips
- Cleft palate or lip
- Examine tonsils and palate
- Respiratory system
- Size and symmetry of thorax
- Small -> pulmonary hypoplasia
- Pectus excavatum or carinatum
- Breathing movements
- Use of accessory breathing muscles
- Auscultation
- Symmetrical, equal breathing sounds
- Size and symmetry of thorax
- Cardiovascular system
- Palpation of peripheral pulses
- Auscultation
- Murmurs in the first few days are normal
- Abdomen
- Slightly protruding abdomen is normal
- Umbilicus
- Look for infection or hernia
- Palpation of liver, spleen, kidneys
- Diastasis recti (nonunion of the two rectus muscles) is common and spontaneously resolves
- Auscultation
- Musculoskeletal
- Sacral dimple
- May indicate underlying neural tube defect
- Structure of spine (kyphosis, lordosis, scoliosis)
- Joints (position, motion, stability, swelling, tenderness)
- Valgus/varus
- Developmental dysplasia of the hip
- Number of digits
- Sacral dimple
- Neurological
- Muscle tone, strength
- Deep tendon reflexes
- Superficial reflexes (abdominal and cremasteric)
- Neonatal (primitive) reflexes
- These are present at birth but resolve after a few months
- They are abnormal if absent in the first months, asymmetric, or persist after the first few months
- Moro reflex (startle reflex)
- When the head suddenly falls back, arms are abducted and extended, and legs are flexed
- Stepping reflex
- Holding infant upright with feet on flat surface elicits a walking movement
- Grasp reflex
- Touching the palm elicits grasping
- Genitourinary
- Presence and description of external genitalia
- Hernias and hydrocoeles
- Presence of cryptorchidism
- Presence of hypospadias and epispadias
- General appearance
5. Birth injuries
It helps to have studied operative vaginal delivery before reading this topic.
Risk factors
-
- Large-for-date infants (Especially > 4500 g)
- Forceps-assisted or vacuum-assisted delivery
- Breech presentation
- Excessive traction during delivery
Extracranial injuries
Cephalohaematoma is a haematoma between skull and periosteum which is a complication of operative vaginal delivery, forceps or vacuum delivery. Because it is a subperiosteal hematoma, it’s limited by the cranial suture lines and don’t cross them. This limits the degree of potential blood loss. These resolve spontaneously within weeks or months, and rarely cause problems.
Subgaleal haematoma is a haematoma in the loose tissue between skull periosteum and epicranial aponeurosis. This is also a possible complication of operative vaginal delivery, especially vacuum delivery. These haematomas are not limited by the suture lines and can therefore grow quite large, often causing haemorrhagic shock. Up to 20 – 40% of neonatal blood volume can be lost. Treatment is supportive (fluid resuscitation or transfusion).
Neurologic injuries
Facial nerve palsy is a potential complication of forceps delivery. It’s a peripheral type of facial nerve palsy. It spontaneously resolves, and so only supportive treatment is necessary. Clinical features include:
- Loss of nasolabial fold
- Partial closing of eye
- “Drooping” mouth
Neonatal brachial plexus palsy is a complication of vaginal birth if excessive traction is applied. This traction stretches the brachial plexus and injures it. These spontaneously resolve in most cases, but in some cases impairment is persistent. Two clinical forms exist:
- Erb’s palsy
- Most common type
- C5, C6 affected
- Adducted, internally rotated arm
- Extended forearm
- Normal hand and wrist movement
- Grasp reflex present
- Klumpke’s palsy
- Rare
- C7 – Th1 affected
- Weakness of hand muscles
- Grasp reflex absent
- Claw hand deformity
6. Hypoxic-ischemic encephalopathy, intracranial haemorrhage, and its complications
Hypoxic-ischaemic encephalopathy
Perinatal asphyxia refers to the condition in which foetal gas exchange is abnormal antepartum, intrapartum, or postpartum, which leads to the neonate’s brain and other organs being deprived of oxygen. It’s characterised by progressive hypoxaemia, hypercarbia, and lactic acidosis. Unless reversed, perinatal asphyxia will cause irreversible CNS damage and other organ damage, or even death.
Hypoxic-ischaemic encephalopathy (HIE) is the name of the CNS complication of perinatal asphyxia. There is a high mortality rate (up to 50%), and possible long-term outcomes of HIE include cerebral palsy, abnormal psychomotor and mental development, and epilepsy.
Etiology
Perinatal asphyxia is caused by a hypoxic or ischaemic event occurring immediately before or during labour. The list of possible causes is long:
- Uterine rupture
- Placental abruption
- Umbilical cord prolapse
- Amniotic fluid embolism
- Vasa praevia
- Foetomaternal haemorrhage
Many risk factors increase the risk of developing perinatal asphyxia in the setting of one of the events listed above. These include risk factors which decrease the adaptability of the mother, placenta, and foetus, including:
- Foetal prematurity, congenital heart defect
- Placental abnormalities
- Maternal diabetes, heart failure, preeclampsia
Clinical features
HIE can be anywhere from mild to severe. In mild cases the neonate may only exhibit poor feeding, irritability, excessive crying, etc., while in severe cases they may be lethergic or comatose, hypotonic or flaccid, have decreased or absent reflexes, seizures, nystagmus, etc.
Diagnosis
Typical clinical features in an infant with signs of perinatal asphyxia (acidosis, low apgar score) suggests the diagnosis. MRI shows evidence of hypoxic-ischaemic brain injury and confirms the diagnosis.
Treatment
All infants with possible perinatal asphyxia or HIE require resuscitation and stabilisation.
Therapeutic hypothermia is the only measure which improves the prognosis of perinatal asphyxia and hypoxic-ischaemic encephalopathy. It refers to reducing the body temperature to 33 – 34°C, which protects the brain and other organs. Therapeutic hypothermia must wait until after resuscitative measures have stabilised the neonate but must be applied no more than 6 hours after birth.
Intracranial haemorrhage
Intracranial haemorrhages are possible birth injuries. The risk is increased in operative vaginal delivery (vacuum or forceps), but it may also occur in unassisted deliveries. It’s most common in prematures. There are four different types. In decreasing order of incidence:
- Subdural haemorrhage
- Subarachnoid haemorrhage
- Intraventricular haemorrhage
- Epidural haemorrhage
The neonatal skull is plastic (the adjective, not the material), and so haematomas can grow relatively large without an increase in intracranial pressure.
Small bleedings may be asymptomatic and incidental findings. Larger bleedings may cause seizures, respiratory depression, or apnoea. Diagnosis is made based on neuroimaging, usually CT in emergent cases and MRI otherwise.
Small bleedings can often be managed conservatively. If they’re large enough to cause intracranial pressure or symptoms, surgical evacuation can be performed. The neonate should be monitored for hypovolaemia and anaemia.
(Respiratory distress)
Respiratory distress is a condition where the neonate has trouble maintaining normal gas exchange and oxygenation, causing a set of typical clinical features:
- Tachypnoea (RR > 60)
- Intercostal and subxiphoid retractions (chest retractions)
- Abdominal breathing
- Expiratory grunting (forced expiration against closed glottis)
- Nasal flaring
- Cyanosis
There are many possible causes of respiratory distress. These are the most common:
- Infant/neonatal respiratory distress syndrome (IRDS/NRDS)
- Transient tachypnoea of the newborn
- Meconium aspiration syndrome
- Pneumonia
- Congenital cyanotic heart disease
- Less common causes:
- Pulmonary air leaks
- Choanal atresia
- Pulmonary hypoplasia
- Diaphragmatic malformation
7. Persistent pulmonary hypertension of the newborns. Patent ductus arteriosus.
Persistent pulmonary hypertension of the newborn (PPHN)
Persistent pulmonary hypertension of the newborn (PPHN) is a disorder characterised by pulmonary hypertension which causes right-to-left shunting which causes severe hypoxaemia. There is no parenchymal lung disease or structural heart disease. The high pulmonary pressure causes right-to-left shunting through foetal circulatory pathways (foramen ovale and ductus arteriosus) in PPHN.
After birth the pulmonary vascular resistance normally decreases. In PPHN abnormalities of the pulmonary vasculature prevent this resistance from decreasing, thereby failing the normal transition from placental circulation to pulmonary circulation which normally occurs after birth, causing pulmonary hypertension.
PPHN mostly occurs in near-term, term, or post-term infants (not in preterms). It’s associated with perinatal asphyxia and meconium-stained fluid.
Clinical features
Symptoms begin within 24 hours of birth and include signs of respiratory distress (tachypnoea, retractions, grunting), and severe cyanosis.
Diagnosis
The diagnosis is suspected in term infants who have severe cyanosis, and is confirmed by echocardiography, which shows signs of pulmonary hypertension and right-to-left shunting (and rules out heart disease). Chest x-ray is usually performed to rule out other pulmonary disorders, but is normal in PPHN.
Because deoxygenated blood is shunted from the pulmonary trunk to the aortic arch through the ductus arteriosus, tissues supplied from arteries originating distally to the aortic arch receive deoxygenated blood, while tissues supplied from arteries originating more proximally receive more oxygenated blood (although still deoxygenated compared to normal because of shunting through foramen ovale). This shunting can be seen clinically when comparing the pulse oximetry reading from either big toe and the right thumb. The right hand is supplied by preductal blood and as such will show higher oxygen saturation than the big toes which are supplied by postductal blood. A difference of > 10 % is characteristic for PPHN.
Treatment
Administering 100% oxygen vasodilates pulmonary vessels and is recommended in all cases. If this is insufficient to maintain normoxaemia, mechanical ventilation or even ECMO may be required.
Inhaled NO also vasodilates pulmonary vessels and may be used for severe cases of PPHN. Sildenafil may also be used.
We should also prevent pulmonary vasoconstriction as much as possible by preventing acidosis and hypercarbia.
It’s important to prevent infection in these already vulnerable infants, and so empiric antibiotics may be used.
Patent ductus arteriosus
The ductus arteriosus is a part of the foetal circulation, as it allows blood to flow from the pulmonary artery to the aorta. It usually closes shortly after birth.
Patent ductus arteriosus (PDA), sometimes called ductus botalli persistens (DBP) in Hungarian literature, refers to the case when the ductus arteriosus fails to close after birth. This causes left-to-right shunting and increased pulmonary blood flow, possibly causing pulmonary hypertension.
The symptoms depend on the amount of extra pulmonary blood flow, which depends on the size of the PDA. Small PDAs are often asymptomatic. Larger ones can cause symptoms of heart failure, like dysponea, cyanosis. Over time an uncorrected large PDA may cause Eisenmenger syndrome.
Physical examination may reveal a continuous machine-like murmur in the left infraclavicular area, as blood flows from the high-pressure aorta into the low-pressure pulmonary trunk throughout the whole heart cycle. Echocardiography confirms the diagnosis.
Most cases of PDA should be managed by closing the vessel. There are two options: pharmacological closure and surgical closure.
Prostaglandins keep the ductus arteriosus open. We can therefore use NSAIDs (prostaglandin inhibitors) like indomethacin to induce closure, especially in preterms.
Surgical closure can be achieved percutaneously with devices which are placed inside the PDA and occlude it. It can also be closed with conventional or minimally invasive surgery where a clip is placed on the vessel.
If the PDA is small and haemodynamically insignificant, we may choose to not close it.
8. Polycythaemia. Hyperviscocity syndrome.
Polycythaemia refers to the condition characterised by venous haematocrit > 65% or haemoglobin > 220 g/L. It’s sort of the opposite of anaemia. Polycythaemia may not cause any clinical features, or it may cause hyperviscosity.
Hyperviscosity syndrome is a syndrome of clinical features which occurs if there’s hyperviscosity due to polycythaemia. The hyperviscosity impairs blood flow, predisposing to ischaemia and formation of microthrombi. Neonatal RBCs are less deformeable than adult RBCs and so are predisposed to this impaired bloodflow. When the hct increases beyond 65% even small increases in hct cause significant increase in viscosity.
Polycythaemia is not uncommon in neonates (1 – 2% of healthy neonates), but hyperviscosity is occurs in approximately 50% of these.
Etiology
- Delayed cord clamping
- Twin-twin transfusion syndrome
- Placental insufficiency
- Maternal diabetes
- Baby born at high altitude
Clinical features
Most polycythaemic infants are asymptomatic. If symptoms occur, these are the most common:
- Cyanosis
- Apnoea
- Vomiting
- Poor feeding
- Hypoglycaemia
Treatment
Asymptomatic neonates with hct 60 – 70% may be treated with IV fluids or simply observed. Asymptomatic neonates with hct > 70% and symptomatic neonates (with any hct) may be treated with partial exchange transfusion.
Partial exchange transfusion (PET) involves removing some of the blood volume and replacing it with fluids. This dilutes the blood, decreasing the haematocrit.
9. Jaundice and hyperbilirubinaemia in the newborn. Kernicterus
Many neonates (1/3) develop jaundice, but in most cases this is physiological and uncomplicated. In neonates, the normal upper limit for total bilirubin depends on the precise age (in hours). Subicterus (also erroneously called scleral icterus) is due to bilirubin deposition in the conjunctiva, and is best visible when total bilirubin exceeds 30 – 50 µmol/L. Jaundice typically develops when the level reaches 250 – 300 µmol/L (15 – 20 mg/dL). The bilirubin level can be measured transcutaneously (POC test) or in blood.
Physiological jaundice
Most infants develop physiological jaundice (physiological unconjugated hyperbilirubinaemia) (total bilirubin >80 µmol/L). Bilirubin levels increase gradually after birth and peak on day 3, never causing jaundice within the first 24 hours. The total bilirubin level does not increase beyond 200 µmol/L in formula-fed or 250 µmol/L in breastfed.
Physiological hyperbilirubinaemia is a result of neonatal RBCs having a short lifespan, and that neonates have impaired bilirubin conjugation and decreased bilirubin excretion.
No treatment is necessary for physiological jaundice as it spontaneously resolves after a few days and never reaches dangerous levels.
Breast milk jaundice
Breast milk-fed neonates may develop jaundice (unconjugated) due to breast milk containing β-glucuronidase, which increases enterohepatic recycling of bilirubin. This rarely causes problems, but temporary cessation of breast feeding may be necessary if the bilirubin level gets very high. Jaundice peaks within two weeks and resolves over the next months.
Breastfeeding jaundice
Breastfeeding jaundice is different from breast milk jaundice. Breastfeeding jaundice is due to insufficient breast milk intake. The abnormally low food intake causes subnormal bowel activity, which impairs bilirubin excretion, causing unconjugated hyperbilirubinaemia. Management includes increasing breastfeeding sessions.
Pathological jaundice
Jaundice in neonates is pathological if any of the following are present:
- Onset within 24 hours of age
- Rapid elevation of bilirubin
- Bilirubin level >300 µmol/L (18 mg/dL)
- Anaemia or hepatosplenomegaly is present
- Jaundice is persisting (lasts > 2 weeks)
- Hyperbilirubinaemia is direct rather than indirect
Etiology
- Conjugated hyperbilirubinaemia
- See topic 10
- Unconjugated hyperbilirubinaemia
- Haemolysis
- Haemolytic disease of the newborn (ABO or RhD incompatibility)
- G6PD deficiency (and other RBC enzyme defects)
- Hereditary spherocytosis (and other RBC cell membrane defects)
- Infection
- No haemolysis
- Polycythaemia
- Haematoma
- Diabetic mother
- Mutation of glucoronyl transferase
- Crigler-Najjar syndrome
- Gilbert syndrome
- Impaired gastric motility (e.g. due to pyloric stenosis)
- Haemolysis
Treatment
Management of pathological hyperbilirubinaemia aims to prevent kernicterus. The decision of when to initiate treatment is beyond the scope of these notes, but we should know the options. Factors which are important include the bilirubin level, whether the infant was preterm, etc. Conjugated bilirubin does not cause toxicity and so conjugated hyperbilirubinaemia is not treated
Phototherapy is usually the first choice. It involves exposing the neonate to blue light which breaks down unconjugated bilirubin into water-soluble products.
Exchange transfusion is usually the second choice. In this procedure the neonate’s blood is exchanged for blood products.
Neonatal bilirubin toxicity
Acute bilirubin encephalopathy (ABE) is the acute manifestation of severe unconjugated hyperbilirubinaemia. It may cause typical features of encephalopathy, including lethargy, hypotonia, fever, seizures, and death. ABE is reversible, but if left untreated it will cause kernicterus.
Kernicterus (chronic bilirubin encephalopathy) is the permanent, long-term complication of severe unconjugated hyperbilirubinaemia. It occurs due to deposition of unconjugated bilirubin (which is lipid-soluble) in the basal ganglia and brain stem. Symptoms include cerebral paresis, hearing loss, gaze palsy, and dental hypoplasia.
Conjugated (direct-reacting) bilirubin does not cross the BBB and so is not toxic.
10. Most common causes of direct hyperbilirubinemia
Direct hyperbilirubinaemia is never physiological. It is not neurotoxic but it’s a sign of serious underlying cholestasis or hepatocellular injury. According to the department, direct hyperbilirubinaemia causes a jaundice which is more greenish than the jaundice in indirect hyperbilirubinaemia.
The following list is for children as a whole.
- Biliary obstruction
- Biliary atresia*
- Choledochal cyst
- Inspissated (thickened) bile due to prolonged haemolysis
- Cholelithiasis
- Tumour
- Infection*
- CMV
- TORCH
- Bacterial infection (UTI, sepsis)
- Genetic/metabolic disorders
- Alagille syndrome*
- Alpha-1 antitrypsin deficiency*
- Cystic fibrosis
- Inborn errors of metabolism (galactosaemia, tyrosinaemia)
- Other
- Parenteral nutrition (hyperalimentation cholestasis)*
- Hypothyroidism
- Drugs
Those marked with an asterisk are among the most common causes in neonates.
11. Neonatal hypoglycaemia, hypocalcaemia
Neonatal hypoglycaemia
Newborns have a tendency to develop transient low blood sugar level physiologically, and there is no specific cut-off value at which blood sugar level in infants is pathological. However, one tacher said hypoglycaemia is < 2,6 mM.
Etiology:
- Due to inadequate glycogen stores
- Prematurity
- IUGR
- Due to impaired glucose production (glycogenolysis or gluconeogenesis)
- Glycogen storage diseases
- Disorders of amino acid metabolism (maple syrup urine disease, etc.)
- Disorders of carbohydrate metabolism (galactosaemia)
- Disorders of fatty acid metabolism
- Due to abnormal glucose homeostasis
- Growth hormone deficiency
- Cortisol deficiency
- Congenital hyperinsulinism
- Other
- Ingestion of antidiabetics, ethanol, beta blockers
- Liver failure
- Sepsis
A hypoglycaemic neonate may exhibit clinical features like lethargy, irritability, tremor, hypotonia, decreased consciousness, and seizures.
We usually don’t routinely screen asymptomatic healthy term infants for hypoglycaemia, but those who are symptomatic or at risk for hypoglycaemia should have their levels checked. Blood glucose can be measured by POC test from capillary blood (which is less accurate) and by regular blood test, which is more accurate and also allows for measurement of other parameters which could provide information about the cause (FFA, lactose, pH, ketones, etc.).
Those at risk include
-
- Preterm and post-term infants
- Large for gestational age (LGA) infants
- Small for gestational age (SGA) infants
- Infants of mothers with diabetes
Treatment involves oral feeding or IV 20% dextrose.
Neonatal hypocalcaemia
Hypocalcaemia is a common problem in neonates. We differentiate early hypocalcaemia (occuring first 2-3 days) from late hypocalcaemia (after 3 days, usually after a week).
Etiology:
-
- Early hypocalcaemia (first two-three days)
- Prematurity
- Maternal diabetes
- Perinatal asphyxia
- Intrauterine growth restriction
- Hypoparathyroidism
- DiGeorge syndrome
- Late hypocalcaemia (day 3 – 7)
- High phosphate intake (cow milk intake)
- Acute kidney injury
- Early hypocalcaemia (first two-three days)
Most affected neonates are asymptomatic, but symptoms include signs of neuromuscular irritability like jitteriness and muscle jerking. In severe cases, seizures and laryngospasm may occur.
Like with hypoglycaemia, only high-risk or symptomatic neonates are screened for hypocalcaemia. Screening is indicated in very low birth weight (VLBW) infants, infants with congenital heart disease, and those with symptoms.
Diagnosis is made by blood test measuring ionised and total Ca2+ level. It may also be useful to measure phosphate, vitamin D, PTH. The limits are:
-
- Total Ca2+ < 2 mM
- Ionized Ca2+ < 1,1 mM
Neonates may be managed with oral feeding or IV calcium gluconate solution depending on the severity.
12. Neonatal convulsions, differential diagnosis, and treatment
Not all seizures or convulsions are epileptic; it’s important to distinguish epileptic neonatal seizures from non-epileptic paroxysmal disorders (non-epileptic seizures). The same pathologies can cause both epileptic and non-epileptic seizures, but the prognosis and treatment are different.
Epileptic neonatal seizures
Epileptic neonatal seizures can be clinical or subclinical, but because they are “true” epileptic seizures there are always EEG changes. It occurs in 1 – 3 / 1000 live birth.
Etiology:
-
- Hypoxic-ischaemic encephalopathy (most common)
- Metabolic disturbance (hypoglycaemia, hypocalcaemia, hyponatraemia, etc.)
- Epilepsy syndromes
- Inborn errors of metabolism
- Ischaemic stroke
- Intracranial haemorrhage
- CNS infection
There are multiple types of seizures. It’s important to know that generalised tonic-clonic (grand mal) seizures never occur in neonates.
- Subtle seizures
- Most common
- Abnormal, rapid eye movements
- Lip smacking
- Swimming or pedalling movement
- Clonic seizures
- Second most common
- Repetitive muscle contractions (2 – 3 per second)
- Tonic seizures
- Sustained, transient muscle contractions
- Myoclonic seizures
- Sporadic (isolated) muscle contractions
- Subclinical seizures
- No clinical signs, just present on EEG
Diagnosis is made by EEG or video-EEG (where continuous video is made of the infant while EEG is taken). Evaluation of blood, CSF, and neuroimaging may be necessary to find the underlying cause.
Treatment involved treatment of the underlying cause. Sometimes antiepileptics are necessary, in which case phenobarbital, phenytoin, or diazepam can be first choices.
Non-epileptic paroxysmal disorders
Non-epileptic paroxysmal disorders are not true epileptic seizures but they may mimic them clinically. They are more common than epileptic neonatal convulsions. They can occur with or without underlying disease
Etiology:
- Benign (idiopathic)
- Metabolic disturbances
- Hypoxic-ischaemic encephalopathy
- Intracranial haemorrhage
- Sepsis
- Drug withdrawal
Clinical features include apnoeic episodes, jitteriness, and benign myoclonus during sleep. These movements can be similar to those of epileptic seizures. There are no EEG changes. Treatment involves treatment of the underlying disease, if present.
13. Transient tachypnoea of the newborn. Meconium aspiration.
Transient tachypnoea of the newborn
After birth, the neonate’s lungs are filled with fluid which is rapidly resorbed and cleared to allow for air to enter the air spaces.
Transient tachypnoea of the newborn (TTN), also called “wet lung disease”, is a lung disorder characterised by delayed resorption and clearance of foetal lung fluid leading to a state of pulmonary oedema. It’s a common cause of respiratory distress in term infants. It’s a benign condition which usually resolves after 12 – 24 hours.
Delivery by C-section is a risk factor for TTN as the uterine contractions a foetus is usually exposed to during vaginal birth appear to help in the resorption of fluid.
The typical presentation is tachyponoea which begins within 2 hours of delivery. There may also be mild/moderate features of respiratory distress, like cyanosis, nasal flaring, retractions, and expiratory grunting.
The diagnosis is clinical, but an x-ray must be performed to rule out other causes of neonatal respiratory distress (NRDS, infection, etc.). In TTN x-ray will show a hyperinflated lung with prominant vascular markings in a sunburst pattern.
Treatment is supportive, as it spontaneously resolves. Oxygen supplementation or noninvasive respiratory support (nasal CPAP or NIPPV) may be required to maintain oxygen saturation until it resolves. Fluid restriction may hasten the resolution.
Meconium aspiration syndrome
Meconium aspiration syndrome (MAS) is a syndrome of respiratory distress and meconium-stained amniotic fluid. The foetus is exposed to and aspirates meconium, which causes pulmonary disease. Meconium inactivates surfactant, obstructs the airway, and causes airway inflammation.
MAS usually affects postterm infants, as they have overmatured GI tract which release meconium before birth.
After birth the neonate is stained by greenish amniotic fluid (from the meconium) and has respiratory distress right after birth.
The diagnosis is made based on clinical findings of meconium-stained amniotic fluid and neonatal respiratory distress as well as typical findings on x-ray. Typical features include hyperinflation of lung, emphysema, and irregular patchy infiltrate. Blood gas shows respiratory failure.
Treatment is generally supportive, ensuring sufficient oxygenation by respiratory management (nasal cannula, noninvasive ventilation, etc.). We may administer empiric antibiotics (ampi+genta). We may perform suction of meconium from the nasopharynx and trachea (controversial).
Because MAS is most common in postterm infants, labour is usually induced at week 41 to decrease the risk of it. Routine intrapartum CTG monitoring can be used to detect and correct foetal hypoxia, which is associated with meconium release.
(Choanal atresia)
Choanal atresia is a congenital malformation with bony or soft tissue obstruction of the choanae. It can be uni- or bilateral. It was included in the lecture but doesn’t have its own topic.
Neonates are obligate nasal breathers, so bilateral choanal atraesia is life-threatening. They develop respiratory distress with intermittent cyanosis, especially when eating and sleeping. Very characteristic is that cyanosis improves when crying, as they can breathe with their mouth when crying. Nasal cannulation and later surgical perforation is required.
Unilateral choanal atraesia usually only causes chronic rhinitis. It can be treated surgically at the age of 1 – 2.
(Air leak syndrome)
Air leak syndrome (pulmonary air leak ) was also included in the lecture but doesn’t have its own topic either. It’s a syndrome which occurs due to air escaping from the lung into spaces where air is no longer present. The most common types are:
- Pneumothorax
- Pneumomediastinum
- Pulmonary intestitial emphysema
- Pneumopericardium
Air leak syndrome is usually a complication of underlying lung disease, like NRDS, mechanical ventilation, and meconium aspiration syndrome.
PTX can cause respiratory distress. Pneumomediastinum or tension pneumothorax can cause respiratory and circulatory collapse. Tension pneumothorax is treated with emergency thoracocentesis and chest tube placement. Physical examination shows shifted heart sounds and chest assymmetry. Pneumomediastinum often spontaneously resolves.
14. Respiratory distress syndrome and its treatment
Infant/neonatal respiratory distress syndrome (IRDS/NRDS), also called hyaline membrane disease (from a pathological standpoint), is a leading cause of preterm neonate morbidity and mortality, and one of the causes of neonatal respiratory distress. The major risk factor is prematurity, and the risk increases as the gestational age decreases:
- < 28 weeks of gestation: > 50%
- > 37 weeks of gestation: < 5%
Pathomechanism
The primary cause of NRDS is pulmonary surfactant deficiency due to an immature lung. This increases surface tension of the alveoli, causing alveoli to collapse, causing atelectasis. This decreases lung compliance and causes V/Q mismatch, which causes hypoxaemia and hypercapnia.
The resulting lung injury causes exudate to leak into the alveoli, forming hyaline membranes.
Clinical features
NRDS presents within the first minutes or hours of life with signs of respiratory distress. Symptoms progress and peak within 3 days, after which they begin to resolve, usually resolving after a week.Diagnosis
Diagnosis
Diagnosis is based on clinical features, prematurity, and x-ray. X-ray shows low lung volume and diffuse ground-glass opacities with air bronchograms. In severe cases we can see a completely opaque lung, so-called “white lung”. Blood gas shows respiratory failure.
The severity of NRDS is based on the Silverman-Anderson scoring. A score of 0 means no respiratory distress, while 10 means the most severe respiratory distress. It’s based on the typical clinical features of respiratory distress (retractions, nasal flaring, grunting).
Treatment
The main treatment is respiratory support and administration of exogenic surfactant. The choice of respiratory support depends on the severity. In less severe cases we can use noninvasive methods like nCPAP or BiPAP, but in severe cases intubation and mechanical ventilation is required.
Administration of exogenous surfactant treats the underlying problem and is performed in many cases. Both natural (better) and synthetic surfactant exist. It is aministered non-invasively by thin catheter after direct laryngoscopy.
Prevention
In case early preterm (< 34 weeks) delivery may be imminent, glucocorticoids (betamethasone or dexamethasone) are administered IM to the mother in two doses, the second dose repeated 24 hours after the first. Glucocorticoids hasten maturation of the foetal lungs (by stimulating surfactant production), reducing the incidence of neonatal respiratory distress syndrome (NRDS) as well as perinatal morbidity and mortality in general. This effect occurs as early as a few hours after the first dose, but the maximum effect is not achieved until 24 hours after the second dose. The effect lasts approximately 7 days.
Tocolytics (beta mimetics, atosiban, nifedipine) may be used to delay labour for a few days, allowing the glucocorticoids to take effect.
Prenatal amniocentesis can assess the foetal lung maturity by measuring the lecithin/sphingomyelin (L/S) ratio in amniotic fluid. If the ratio is < 1,5 there is a high risk for NRDS.
15. Nosocomial infections in neonates (pathogens, treatment, prevention)
Nosocomial neonatal infections are all neonatal infections where there is no evidence of transplacental transmission. The most common infections are ventilator-associated pneumonia, central line-associated bloodstream infection (CLABSI), urinary tract infection, and sepsis.
These more frequently occur in neonates with risk factors like:
- Prematurity
- Very low birth weight
- Endotracheal tube
- Admission into neonatal ICU
- Central catheter
- Indwelling urinary catheter
- Broad-spectrum antibiotic use
- CNS shunt
The most common pathogen is coagulase-negative staphylococci (CONS), which accounts for 70% of cases. The most common CONS is staphylococcus epidermidis. Other potential pathogens include staphylococcus aureus, gram negative rods (E. coli), etc.
In all neonates with evidence of infection (fever/hypothermia) a full diagnostic workup is indicated, including blood culture, urine culture, and CSF culture. We administer empiric antibiotics until the culture result is available, usually ampicillin + gentamycin/cefotaxime.
Preventative measures include:
- Hand hygiene
- Maternal breast milk feeding
- Correct aseptic technique when inserting and maintaining catheters, tubes
- Avoid broad-spectrum antibiotics
16. Clinical symptoms, diagnostic features, and treatment of sepsis in the newborn period
Neonatal sepsis is a severe condition with high morbidity and mortality, which is the reason why all signs of infection in neonates warrant throrough evaluation and urgent empiric antibiotic treatment. There are two types of neonatal sepsis, early-onset sepsis and late-onset sepsis.
Early-onset occurs in the first week, often the first day. It accounts for 3/4 of neonatal sepsis cases, and is often fulminant, multisystem, and severe, with a mortality rate up to 50%. Early-onset sepsis often begins in utero.
Late-onset occurs after the first week, often in weeks 3 – 4. It progresses more slowly, affects a single or a few organ systems, and has a relatively lower (but still high) mortality rate of up to 20%.
Neonatal sepsis, pneumonia, UTI, and meningitis present similarly, have similar findings, and treatment. For these reasons, they are sometimes collectively called neonatal invasive disease.
Risk factors
- Prematurity (5x higher)
- Maternal colonization by GBS (see topic B6 of obgyn 1)
- Traumatic delivery
- Prolonged rupture of membranes
Microbiology
The most common causative pathogen of early-onset neonatal sepsis and other neonatal infections is Group B streptococcus (GBS). Many pregnant women are colonised by GBS which can infect the neonate. The other three most common microbes are E. coli, Listeria, and Klebsiella. This is important to know, and you can remember it by there being two gram positives and two gram negatives.
The most common causative pathogens of late-onset disease are CONS, staphylococcus aureus, etc.
Clinical features
Typical clinical features include respiratory distress, temperature instability (fever/hypothermia), lethargy, poor feeding, etc. In practice it’s difficult to differentiate sepsis from other common disorders like NRDS and perinatal asphyxia based on clinical features.
Diagnosis
Neonatal sepsis is diagnosed when there are clinical features of systemic infection and positive blood/CSF culture. There may be leukocytosis or leukocytopaenia and elevated CRP and PCT.
Treatment
Empiric antibiotics are initiated in case of clinical suspicion, preferably after cultures are made. The choice is usually ampicillin plus either gentamicin or cefotaxime. GBS is sensitive to penicillin G.
Prevention
Screening and eradication of GBS (with antibiotics) in colonised pregnant women is important in preventing neonatal sepsis (see topic B6 of obgyn 1).
17. Meningitis in the newborn period
Bacterial meningitis
Bacterial sepsis and meningitis are closely linked in neonates, as most cases of neonatal meningitis are caused by haematogenous spreading to the CNS during sepsis. The symptoms of both can be difficult to differentiate clinically, but they have similar risk factors, features, microbiology, and management. Bacterial meningitis is more common in the first month than at any other time in life.
Neonatal meningitis is a severe condition with poor prognosis due to high risk of permanent neurological complications and death.
Like sepsis, we also distinguish early-onset (first week) and late-onset (later) bacterial meningitis.
Clinical features are similar as for sepsis, with the addition of irritability and seizures. Neck stiffness is rare in neonatal meningitis.
As part of the standard evaluation of sick neonate, blood, CSF, and urine cultures are obligatory. Findings are similar to those of sepsis, with the addition of the following CSF findings:
- Leukocytosis (> 20 WBCs/µL)
- Elevated proteins (> 2 g/L)
- Decreased glucose
- Positive gram staining
- Positive CSF culture
Empiric treatment is usually ampicillin plus either gentamicin or cefotaxime.
HSV meningitis
HSV meningitis is not frequent in newborns, but it may occur. It causes similar clinical features as bacterial meningitis with the possible addition of vesicles or HSV-related birth defects (microcephaly, microophthalmia). Empiric acyclovir therapy is initiated upon clinical suspicion. PCR of vesicles and CSF gives the diagnosis.
18. Neonatal necrotizing enterocolitis
Necrotising enterocolitis (NEC) is the most common GI emergency in neonates. It mostly affects preterms (especially those < 28 weeks) in their 2 – 3 week after previously having appeared healthy.
Pathomechanism
The pathomechanism is not well known, but it involves bowel ischaemia and GI infection/dysbiosis.
Clinical features
NEC presents with sudden changes in the condition, including poor feeding and nonspecific signs like respiratory failure, lethargy, apnoea, etc. Characteristic features include a distended and shiny abdomen, diarrhoea (especially bloody), and bilious vomiting.
In later stages DIC, sepsis, and shock can develop. Discoloured (erythematous) abdominal wall is a late sign.
Diagnosis
Gastrointestinal air leak is the keyword here. X-ray or ultrasound may show air in the bowel wall, air in the hepatobiliary system, or free air in the abdomen. The diagnosis is made when there are both clinical and radiological features. Blood test may show thrombocytopaenia and metabolic acidosis.
Treatment
Treatment involves the following:
- Stabilisation (especially fluids)
- Nil per os (total peripheral nutrition)
- Gastric decompression with a gastric tube
- Antibiotics (ampicillin + gentamicin + metronidazole)
In case of GI perforation, severe peritonitis or suspected necrotic bowel, surgery is necessary.
Prevention
Feeding high-risk neonates (mostly extremely preterms) breast milk rather than formula, as well as administering probiotics reduce the risk for developing NEC.
19. Congenital and connatal infections
General
Regarding these infections, it’s important to know:
- The nature of the damage to the foetus
- The treatment, if available
- Preventative measures, if available
For antenatal screening of congenital infections, see topic B6 in obgyn 1.
Most congenital infections cause nonspecific symptoms, like:
-
- Petechiae, purpurae
- Hepatosplenomegaly
- Small for gestational age/IUGR
- CNS abnormalities (microcephaly, calcifications)
- Seizures
- Haematological abnormalities (thrombocytopaenia, haemolytic anaemia)
- Pneumonia
- Miscarriage/stillbirth
Congenital rubella infection
Congenital rubella infection is very rare in countries with vaccination programmes containing the MMR vaccine. There can be both early and late manifestations.
- Early manifestations
- IUGR
- Hearing loss
- CNS involvement (infection, microcephaly)
- Congenital heart disease
- Eye problems (cataract, glaucoma)
- Late manifestations
- Diabetes mellitus
- Thyroid disease
- Eye problems (cataract, glaucoma)
- Delayed psychomotor development
Congenital rubella is effectively prevented by vaccination programmes containing the MMR vaccine.
Congenital cytomegalovirus infection
Congenital CMV infection is among the most common congenital infections, and a leading cause of hearing loss worldwide. 90% of neonates are asymptomatic at birth.
The most common early manifestations are non-specific and include petechiae, jaundice, hepatosplenomegaly, IUGR, etc. Possible late manifestations include hearing loss, vision loss, dental abnormalities, delayed psychomotor development, etc. Hearing loss is the most common finding.
The diagnosis can be made with viral culture or PCR of urine or saliva. No prevention is available for congenital CMV. Infants with proven CMV infection should be treated with IV ganciclovir or vangalciclovir.
Congenital toxoplasmosis
Cats are the final hosts of toxoplasma gondii. Parasite cysts or eggs are found in cat faeces, which can contaminate soil, dirt, and sand. The eggs can also be ingested by other animals, forming cysts in their muscles which may end up on a plate as meat to eat.
Congenital infection may cause a classical triad of hydrocephalus, chorioretinitis (posterior uveitis), and intracranial calcifications. It may also cause non-specific features like hepatosplenomegaly, thrombocytopaenia, and IUGR.
Maternal toxoplasmosis can be avoided by the following:
- Avoid raw meat, meat not properly heated, and unpasteurised milk
- Vegetables and fruit should be properly washed
- Avoid work with soil, dirt, and sand
- Avoiding handling cat faeces
Maternal infection is managed with antibiotics. The exact choice depends on local guidelines. The following guidelines are Norwegian (I couldn’t find Hungarian ones):
- Foetal infection not (yet) discovered or gestational week < 14: azithromycin or spiramycin
- Foetal infection documented and gestational week > 14: Combination therapy with pyrimethamine + sulphadiazine + folinic acid
Pyrimethamine and sulphadiazine are teratogenic in the first trimester and therefore not given until approx. week 14.
Congenital syphilis
Syphilis is always the worst infectious disease to study because it can cause virtually any complication:
-
- Early manifestations
- Miscarriage/stillbirth
- Fever
- Hepatomegaly
- Rhinitis
- Rash
- +++
- Late manifestations (> 2 years of age)
- Saddle nose
- Prominent forehead
- Hutchinson teeth (notched, widely spaced teeth)
- Mulberry molars (poorly developed first molars)
- Hearing loss
- Gummas
- Early manifestations
Pregnant women are routinely screened for syphilis antenatally. Antibiotics are indicated for all pregnant women with positive serology and confirmatory test, unless they can document previous successful treatment. The treatment is the same for pregnant women and newborns with congenital infection, 10 days of IV penicillin G or single-dose IM penicillin G for pregnant women and newborns.
Congenital listeriosis
Congenital listeriosis can cause:
- Miscarriage/stillbirth
- Granulomatosis infantiseptica (widespread abscesses in multiple internal organs)
- Neonatal meningitis or sepsis
Pregnant women should avoid soft cheeses and meats. Congenital infection is treated with ampicillin and gentamicin.
Congenital parvovirus B19
Clinical features in foetus:
-
- Miscarriage/stillbirth
- Severe anaemia
- Foetal hydrops
Treatment of foetal anaemia is intrauterine blood transfusion. Prevention includes proper hand hygiene.
Congenital varicella zoster
Congenital varicella zoster is rare thanks to the VZV vaccine. Clinical features in foetus:
- Congenital varicella syndrome (infection during first 20 weeks)
-
- Hypertrophic scars
- Limb abnormalities
- Eye abnormalities
- CNS abnormalities
- Neonatal varicella (infection right before delivery)
- Severe infection
- Mortality rate 30%
-
Prevention and treatment:
-
-
- IV acyclovir for infected pregnant women and neonates
- If maternal symptoms right before delivery: administer anti-VZV immunoglobulins
-
Congenital herpes simplex
Congenital HSV infection (usually HSV2) occurs after intrapartum exposure to genital herpes. It can cause typical vesicular skin lesions and herpes simplex encephalitis in the neonate, which is a serious condition.
To prevent congenital HSV, delivery is made by C-section if the mother has active genital herpes symptoms at the time of delivery. The treatment is acyclovir.
Hepatitis B
Hepatitis B can be transmitted transplacentally or during birth. Maternal antenatal screening for HBV is routine. In case of an HBV positive mother she may receive antivirals, and the neonate is given both HBV vaccine and anti-HBV immunoglobulin.
Hepatitis C
Hepatitis C can be transmitted during birth. There’s nothing to do to decrease the risk of transmission, and there’s no treatment for infected children. Ideally, hepatitis C positive women should undergo HCV eradication therapy before attempting pregnancy.
HIV
HIV can be transmitted during birth and breastfeeding. Maternal HIV is treated with HAART during the whole pregnancy. If the viral load is high, delivery by C-section is recommended. The neonate should receive postexposure prophylaxis (zidovudine, AZT) after birth.
Chlamydia trachomatis
Chlamydia trachomatis may be transmitted during birth and cause neonatal conjunctivits and pneumonia. Screening pregnant women for chlamydia is important to prevent it. Neonatal infection is treated with oral azithromycin.
20. Inborn errors of metabolism
Inborn errors of metabolism disorders characterised by deficiencies in enzymes involved in the metabolism. They’re classified according to the part of the metabolism they involve:
- Disorders of amino acid metabolism
- Phenylketonuria
- Homocystinuria
- Maple syrup urine disease
- +++
- Disorders of carbohydrate metabolism
- Galactosaemia
- Glycogen storage disorders
- Fructose intolerance
Many of these disorders are screened for during routine neonatal screening, and so they’re often diagnosed before they can cause complications.
Phenylketonuria (PKU)
Phenylketonuria (PKU) is characterised by inability to convert phenylalanine to tyrosine. There are two types of PKU, classic and atypical, depending on the degree of enzyme deficiency. Classical PKU accounts for 98% of cases and is characterised by complete enzyme deficiency of phenylalanine hydroxylase.
Accumulation of phenylalanine causes intellectual disabilty and abnormal brain development, and tyrosine deficiency causes a deficiency of catecholamines, melanin and thyroxine, which are derived from tyrosine.
Thanks to neonatal screening, infants rarely develop complications from PKU these days. On day 2/3 the level of serum phenylalanine is measured (as the neonate must be fed phenylalanine before abnormal levels could be detected). The diagnosis is confirmed by genetic testing.
Possible complications include intellectual disability, epilepsy, and light pigmentation.
The treatment is life-long dietary restriction of phenylalanine (or in the future, ezyme therapy). This prevents complications of PKU. Notably, the artificial sweetener aspartame contains phenylalanine.
Galactosaemia
Galactosaemia is characterised by deficiency of enzymes involved in galactose metabolism. Different enzymes can be deficient, but the most common is deficiency of galactose 1-phosphate uridyltransferase (GALT), which causes 90% of cases and is known as classic galactosaemia.
Screening for galactosaemia is also routine and involves measuring serum levels of galactose and its metabolites on day 2/3. The diagnosis is confirmed by enzyme assay of RBCs, which measure the enzyme activity.
The typical presentation of untreated galactosaemia is jaundice, vomiting, hepatomegaly, and E. coli sepsis.
Breast milk and regular (cow’s milk-based) formula contains galactose, so affected infants must be fed soy milk-based formula and avoid galactose-containing foods for life. Calcium supplementation is recommended to prevent deficiency.
Unfortunately, despite adherence to dietary restriction, neurodevelopmental and neuropsychological problems may develop.
Glycogen storage disorders
Glycogen storage disorders (GSD) are disorders characterised by abnormal storage of glycogen due to defects in enzymes of glycogenolysis or glycolysis. There are many different types with different clinical features, and the age of onset varies from birth to adulthood.
Common for all GSDs is that glycogen accumulates, and because glycogen is stored in the liver, heart, and muscle, the problems occur there. Most present during infancy or childhood. These are the most common types:
- GSD type I – von Gierke disease
- GSD type II – Pompe disease
- GSD type III – Cori disease
- GSD type IV – Andersen disease
- GSD type V – McArdle disease
The most common manifestations of glycogen storage disorders are hypoglycaemia, hepatomegaly, muscle pain, exercise intolerance, and cardiomyopathy. Symptoms often improve after eating or glucose administration.
Biopsy of muscle or liver shows excessive glycogen storage. Enzyme assays can be used to measure enzyme activity. Genetic testing can confirm the diagnosis.
Most GSDs can be managed effectively with dietary therapy, for example uncooked cornstarch and glucose preparations, although enzyme therapy exists for some types.
Fructose intolerance
Fructose intolerance is similar to galactosemia. When fructose is ingested, deficiency of fructose-1-phosphate aldolase leads to accumulation of fructose 1-phosphate. This causes vomiting, hypoglycemia, and severe liver and kidney disease. The treatment is elimination of fructose and sucrose from the diet, which prevents clinical disease.
Fructosuria is an asymptomatic analogue of fructose intolerance. It’s caused by fructokinase deficiency, but it doesn’t cause any clinical consequences other than asymptomatic fructosuria.
21. Late complications of neonatal diseases (ROP, BPD)
Retinopathy of prematurity
Retinopathy of prematurity (ROP) is a complication of oxygen toxicity in premature infants. It may cause permanent visual problems or blindness, and it’s a leading cause of blindness in very low birth weight premature infants.
Excessive arterial oxygen tension causes vasoconstriction of immature retinal arteries, causing the vessels to be obliterated. Term infants are not susceptible as their retina is completely vascularised.
Very low birth weight infants (< 1500g) and very premature (< 28 – 32 weeks) should be screened for visual problems when they’re 4 weeks old.
To reduce the risk of ROP, VLBW and very premature infants receiving oxygen therapy should have an arterial oxygen tension of no more than 70 mmHg. This doesn’t eliminate the risk, but reduces it considerably.
Most cases of ROP spontaneously resolve, but high-risk ROP should be treated with laser photocoagulation or intravitreal injection of an anti-VEGF drug to reduce the risk for permanent complications.
Bronchopulmonary dysplasia
Bronchopulmonary dysplasia (BPD) is a form of chronic lung disease which develops in preterm infants who have received supplemental oxygen or mechanical ventilation for another cause, most commonly for NRDS. It’s a result of damage to the lungs from this artificial ventilation. Most affected infants are very premature (< 28 – 32) or VLBW.
BPD presents as failure of respiratory distress to improve after 28 days of age (or 36 weeks postmenstrual age), making these infants still dependent on oxygen supplementation at this point.
The oxygen reduction test may assist in the diagnosis. In this test, oxygen support is removed for 60 minutes. If the O2 saturation falls below 90% within 60 minutes of being in room air the test is positive. On x-ray, BPD is characterised by diffuse haziness and coarse interstitial pattern.
Most infants with BPD improve gradually over the next months. Those with severe BPD may develop pulmonary hypertension and cor pulmonale.
Treatment is supportive. It’s important to ensure proper food intake. Fluid restriction is often used. In severe cases, bronchodilators or glucocorticoid may be used.
To reduce the risk of BPD in those who require ventilatory support, we should use as gentle and as little ventilatory support as possible. The target SpO2 should not be higher than 95%. Measures which reduce the risk for NRDS (like antenatal glucocorticoids) indirectly reduce the risk of BPD. Prophylactic IV caffeine therapy should be used routinely in extremely preterm infants to prevent apnoea.
22. Congenital anomalies of the gastrointestinal tract (oesophagus, stomach, intestines)
Oesophageal atresia
Oesophageal atresia is the condition where the oesophagus ends blindly instead of leading to the stomach. There are multiple different types, but type C accounts for 90% of cases and is characterised by a tracheoesophageal fistula which connects to the distal segment of the oesophagus.
Because the foetus cannot swallow amniotic fluid, polyhydramnios develops. After birth, air enters the stomach through the fistula, causing gastric distension. Gastric secretions can enter the lung through the fistula, causing aspiration pneumonia.
The disorder presents immediately after birth with excessive secretions that cause drooling, choking, respiratory distress, and inability to feed. Inability to pass a feeding tube or gastric distension should raise suspicion. X-ray after attempted feeding tube placement shows that the tube is curled in the upper oesophageal pouch.
Tracheoesophageal fistula is associated with other abnormalities of the mesoderm. These abnormalities are called VACTERL:
- 50 – 70% of patients have one or more of these
- Vertebral anomaly
- Anorectal malformations
- Cardiac anomaly
- Tracheoesophageal fistula
- Esophageal atresia
- Renal anomaly
- Limb malformation
Infants with tracheoesophageal fistula should be evaluated for other VACTERL abnormalities.
The definite treatment is surgery, which ligates the fistula and forms an anastomosis of the oesophageal segments. Until surgery can be performed, it’s important to disallow oral feeding and perform continuous suction of the proximal oesophagus to prevent aspiration. The prognosis is very good nowadays.
Duodenal atresia and stenosis
Duodenal atresia refers to complete occlusion or absence of the lumen, while duodenal stenosis refers to narrowin of the lumen. It’s highly associated with Down syndrome, and 30% of duodenal atresia/stenosis cases occur in trisomy 21. The obstruction can occur at any level of the duodenum.
Due to the GI obstruction, polyhydramnios occurs. If the obstruction is distal to the Vater papilla, there may be bilious vomiting. Other symptoms include vomiting, dehydration, and distended upper abdomen.
The “double bubble” sign is characteristic. It refers to the presence of two air bubbles, one being stomach and the other being the distended duodenum proximal to the obstruction, and can be visualised antenatally on ultrasound or postnatally on X-ray. The bowel distal to the obstruction contains no gas.
The definite treatment is surgery, which forms a bypass. Until surgery can be performed, it’s important to disallow oral feeding and perform continuous suction of the stomach (gastric decompression).
Small bowel atresia and stenosis
Small bowel atresia and stenosis are less common than the previous malformations. Symptoms include bilious vomiting and abdominal distension, and x-ray shows distended air-filled bowels proximal to the obstruction. Treatment is similar as for duodenal atresia.
(Anorectal malformations)
Not a topic, but may be important?
Anorectal malformations are those defects characterised by the absence of a normal anal opening. There may be a fistula to the perineum, vaginal vestibule, urethra, or there may be a common opening for the rectum, urethra, and vagina called a “cloaca”.
For evaluation, a lateral x-ray is made 18 hours after birth (as we must wait for air to reach the rectum. We should look for associated VACTERL malformations. The lower (more distal) the malformation, the better the prognosis. The treatment is surgery.
23. Omphalocele, gastroschisis
Abdominal wall defects and treatment
Abdominal wall defects are birth defects characterised by abdominal contents herniating through the abdominal wall. There are two types, omphalocoele and gastroschisis. Fluid loss is rapid as long as the abdominal organs are exposed to air.
The treatment of both types is similar. Preoperatively the herniated abdominal contents are wrapped with sterile saline dressings and covered in plastic wrap to minimise fluid loss. A nasogastric tube is placed to decompress the bowels. Supportive therapy (fluids, normothermia) is important.
The definitive treatment is surgery, during which the contents are returned into the abdominal cavity, and the abdominal wall defect sutured. If any bowels can’t fit in abdominal cavity during surgery, they’re covered by a plastic bag called a silo. The other end of the silo is hung up, so that the bowels will return into the abdominal cavity after a few days due to gravity.
Omphalocoele
Omphalocoele is a result of failure of the midgut to return to the abdominal cavity after physiological herniation. The abdominal organs herniate through abdominal wall through the umbilicus, and the organs are covered by peritoneum and amniotic membranes.
Omphalocoele is associated with other congenital anomalies, especially cardiac defects, malrotation of the intestines, and Beckwith-Wiedemann syndrome. The abdominal wall defect is larger than in gastroschisis.
Gastroschisis
Gastroschisis is a result of the peritoneal cavity being too small for the developing abdominal organs, causing the anterior abdominal wall to rupture at its weakest point, to the right of the umbilicus. The abdominal contents are not covered by peritoneum or membranes. Gastroschisis is not associated with other congenital anomalies, except for intestinal atresia.
24. Diaphragmatic hernia
Congenital diaphragmatic hernia (CDH) is a relatively common birth defect. Impaired development of pleuroperitoneal membrane causes a defect in the diaphragm, 90% of cases occur on the left side. Abdominal contents herniate into pleural cavity, causing the lung to get compressed in utero. This leads to pulmonary hypoplasia, as the lung cannot expand, which is the main complication of CDH.
There are two types, Bochdalek hernia, which is the most common and occurs on the back, and Morgagni hernia, which occurs on the lateral side and is rare.
Bowel in chest produces no symptoms. All clinical features are a result of pulmonary hyperplasia, which presents in the first hours or days of life as respiratory distress.
Bowel sounds and absent breathing sounds on the left side are characteristic, and the diagnosis is confirmed when x-ray shows bowels in the thoracic cavity.
The surgery itself is simple, the problem is stabilizing the neonate first. Mask ventilation should never be used, as it forces air into the intestines, which further compresses the lung. The infant should be ventilated by endotracheal tube intubation.
Surgery can wait up to approx. 10 days and involved repositioning of the herniated organs and simple primary closure of the diaphragmatic defect, sometimes with a patch if the defect is big.
25. Acyanotic congenital heart diseases
Pre- and postnatal circulation
In the prenatal circulation both ventricles work in parallel helping each other, and as such a problem with one side of the heart can be compensated by the other.
In the postnatal circulation the ventricles work in series, and so a problem with one side can’t be compensated and will affect the other as well. For this reason, most congenital heart diseases only become apparent after birth.
Acyanotic congenital heart diseases introduction
These are the congenital heart diseases (CHDs) characterised by left-to-right shunting and an absence of cyanosis.
Many cases of congenital heart disease are associated with factors like trisomies (especially 21), maternal infections, and maternal teratogens (drugs/alcohol).
The most common acyanotic congenital heart diseases are
-
- Ventricular septal defect (VSD)
- Atrial septal defect (ASD)
- Patent ductus arteriosus (PDA)
- AV canal
These CHDs don’t cause cyanosis, but can cause symptoms like exercise intolerance, growth restriction, and heart failure. If severe and leaft untreated, acyanotic CHDs can cause Eisenmenger syndrome.
Physiologically, the pulmonary vascular resistance decreases during months 2 – 3 as the pulmonary vasculature matures. Because of this, some acyanotic CHDs worsen during this time, as the decreased resistance increases the amount of shunting.
Heart defects are evaluated and diagnosed by echocardiography. Smaller defects are often asymptomatic and don’t require treatment.
Ventricular septal defect
Ventricular septal defect (VSD) is the most common CHD overall. It may occur alone or in combination with other defects (like Fallot).
Small defects are asymptomatic. Larger defects cause heart failure after 2 – 3 months.
On physical examination a holosystolic murmur can be heard on the left lower sternal border. There may also be a mid-diastolic murmur at the apex of the heart due to increase transmitral flow. Echocardiography gives the diagnosis.
1/3 of defects close spontaneously. Small defects which don’t close spontaneously but are asymptomatic may not require surgery. Surgery is indicated for symptomatic defects, and is performed at 3 – 6 months. While waiting for surgery, heart failure treatment may be necessary.
Atrial septal defect
Atrial septal defect (ASD) is the second most common CHD. The most common type is the ostium secundum type, in which the defect is in the region of the foramen ovale.
Small defects are asymptomatic. Very large defects cause heart failure after 2 – 3 months, but even large defects are often asymptomatic in childhood. However, symptoms like exertional dyspnoea and fatigue may present in adulthood.
On physical examination a widely split second heart sound which does not change with respiration (it’s fixed) can be heard, as well as a systolic ejection murmur over second ICS on left side. Echocardiography gives the diagnosis.
Treatment is by percutaneous or surgical closure at the age of 3 – 5.
Other acyanotic CHDs
- Patent ductus arteriosus (PDA) – covered in topic 7
- Endocardial cushion defect
- Coarctation of aorta
- Pulmonary valve stenosis
26. Cyanotic congenital heart diseases
Cyanotic congenital heart diseases are those congenitla heart diseases which cause right-to-left shunting, which results in cyanosis and hypoxaemia.
Many cases of congenital heart disease are associated with factors like trisomies (especially 21), maternal infections, and maternal teratogens (drugs/alcohol).
There are 7 major types (the 5 Ts + a few extra):
- Transposition of the great vessels
- Tetralogy of Fallot
- Truncus arteriosus
- Tricuspid atresia
- Total anomalous pulmonary venous connection
- Pulmonary atresia
- Hypoplastic left heart syndrome
We’ll focus on the first two.
Some cyanotic CHDs are so-called duct dependent, meaning that they’re only compatible with life as long as a left-to-right shunt like the ductus arteriosus or VSD persists. For this reason, infants with duct dependent CHD require continuous prostaglandin E infusion to prevent the ductus arteriosus from closing. Transposition of the great arteries, pulmonary atresia, and tetralogy of Fallot (with severe RBOT) are duct dependent.
Tetralogy of Fallot
Tetralogy of Fallot (TOF) is the most common cyanotic CHD. It includes:
- Right ventricular outflow tract obstruction (RVOTO)
- Right ventricular hypertrophy
- Ventricular septal defect
- Overriding aorta
Infants with TOF have mild cyanosis continuously, although cyanosis might not be easily visible in the first two months. During periods of stress, the infant can develop hypercyanosis and hypoxaemia. These episodes are called “tet spells”. The child prefers to sit in a squatting position as this position reduces the right-to-left shunt.
TOF can be diagnosed antenatally with ultrasound or postnatally with echocardiography. A loud harsh ejection systolic murmur can be heard at the left sternal edge from day 1 of life. X-ray shows a characteristic “boot-shaped” heart. Nowadays most are diagnosed antenatally or during routine neonatal checkup, before cyanosis develops.
Medical therapy for heart failure is used until 6 months of age, at which surgery is performed to close the VSD and relieve the RVOTO. If the RVOTO is severe the infant requires prostaglandin E infusion. During tet spells, sedation (morphine), propranolol and muscle paralysis with artificial ventilation may be necessary, but they’re often self-limiting.
Transposition of the great vessels
Transposition of the great vessels (or great arteries) refers to the situation when aorta arises from right ventricle and the pulmonary trunk from the left. This is a ductal-dependent CHD, as the ductus arteriosus is the only thing which mixes the pulmonary and systemic circulations in this case.
Cyanosis is always present following day 2 of life, when the ductus arteriosus closes, and may be severe and life-threatening. If there’s an ASD or VSD, symptoms may be less severe or present later.
It can be diagnosed antenatally with ultrasound or postnatally with echocardiography. X-ray shows a characteristic “egg on a string” appearance of the heart. There are usually no murmurs, but the second heart sound can be louder than normal.
The infant always requires prostaglandin E infusion. If the ductus arteriosus is insufficient, we may make a temporary septostomy in the atrial septum. Surgery to switch the two great vessels (arterial switch procedure) is necessary and can be performed within the first few days.
27. Infant nutrition. Comparison of breastfeeding and formula feeding. Feeding of the premature babies
Infant nutrition
Infants require a lot of nutrients to sustain their rapid growth. The ideal food for infants is breast milk. Ideally, the infant should be exclusively breastfed during the first 6 months, and partially breastfed for the next 6 months, with vegetables and fruits added to the diet to supplement even more nutrients.
There are very few medical contraindications to breastfeeding:
-
- Maternal HIV
- Maternal active TB
- Maternal cytostatic treatment
- Infantile galactosaemia
As such, less than 1% of infants have a medical reason to not be exclusively breastfed.
Unfortunately, after the introduction of infant formula, the level of exclusive breastfeeding in the first 6 months is much lower than ideal. This is likely due to factors like maternal embarrassment, physical breast problems (soreness, pain), and difficulty with combining breastfeeding with work.
In Hungary, only approx. 60% are exclusively breastfed during the first month, and this number decreases with age. Only approx. 40% are exclusively breastfed in the first sixth months. These numbers are better in Scandinavia and Germany.
Breast milk contains enough energy and nutrients for the infant for the first 6 months, but after this the infant needs additional food (fruits and vegetables) to cover their needs.
Exclusively breastfed infants should daily vitamin D supplementation (400 IU) from birth, as breast milk doesn’t contain enough of this vitamin. They should also receive iron supplement from 4 – 6 months, as breastmilk doesn’t cover the iron requirements at this time.
Cow’s milk should not be introduced within the first 12 months of age, as it can lead to iron deficiency due to low iron contect.
History of development of breast milk replacement
Many replacements for breast milk were tried in the 1800s. Cow’s milk was the obvious choice, but infants died days after being fed cow’s milk. It turned out that cow’s milk has 3x the protein content of breast milk, which is problematic. They tried diluting cow’s milk 3x, but this solution didn’t contain enough energy. 3x diluted cow’s milk with added carbohydrates worked as a replacement for breast milk and became the standard substitution.
Formula feeding
The composition of formulas can’t be exactly equal to that of breast milk and will always be inferior.
Infant formula is still based on cow’s milk, and therefore contains cow’s milk proteins. These proteins can be recognized as foreign by the infant and lead to cow’s milk allergy or intolerance. Enzymatic hydrolysis of the cow’s milk protein breaks down epitopes, preventing the infant from recognizing them, but creates an unpalatable formula. For this reason, formulas are only partially hydrolysed, leaving some epitopes.
It’s difficult to mimic the fat composition of breast milk, especially the composition of polyunsaturated fatty acids. Formulas (sold in Europe) must have essential polyunsaturated fatty acids added. Many formulas contain probiotics or prebiotics.
Comparison between breastfeeding and formula feeding
- Pros vs formula feeding
- Pros for the infant
- Contains oligosaccharides which help develop a healthy intestinal bacterial flora
- Contains IgA antibodies, complement factors, and lactoferrin (antimicrobial protein) -> passive immunity
- Contains proteins which increase iron absorption
- Skin-to-skin contact
- Prevents acute infections (like otitis media, pneumonia)
- Lower risk of asthma, allergy, obesity, diabetes mellitus, IBD
- Pros for the mother
- Faster uterus involution
- Longer postpartum anovulation
- Faster weight loss back to baseline
- Lower risk of breast cancer, endometrial cancer, ovarian cancer, type 2 diabetes
- Cheaper, more practical
- Pros for the infant
The intestinal flora of breastfed infants has mostly bifidobacteria, lactobacillus, which is beneficial. The intestinal flora of formula-fed infants consists of mostly E. coli, which is less so.
| Breast-feeding | Formula feeding | |
| Nutrient composition | Physiological | Non-physiological |
| Maintenance of sterility | Easy | Difficult |
| Protective immunity | IgA, complement factors, lactoferrin | None |
| Allergic proteins | None | Cow milk/soy |
| Resulting intestinal flora | Lactobacillus, bifidobacteria | E. coli and coliform strains |
| Mother-infant relation | Very close, skin-to-skin contact | Impersonal |
| Taste | Variable | Always the same |
| Cost | Very cheap | Expensive |
| Practicality | Very practical | Impractical |
Feeding of premature infants
Unlike term infants, preterm infants have no energy store and require nutrients immediately after birth. They also have a higher need for energy and nutrients.
Infants < 32 weeks or < 1800 g require at least one month of a special nutrition plan consisting of initial parenteral feeding which is gradually decreased as enteral feeds increase:
- Six steps of feeding premature infants
- 1 – Parenteral glucose and electrolytes
- Immediately after birth
- 2 – Add parenteral amino acids and vitamin
- ASAP, but within 48 hours
- 3 – Add parenteral lipids
- After 2 days
- 4 – Add minimal enteral nutrition
- Only 12 – 24 mL of breast milk daily
- Stimulates the GI tract function and development
- 5 – Combine parenteral and enteral nutrition
- Lasts for several weeks
- 6 – Only enteral nutrition
- Very variable when this stage is reached
- 1 – Parenteral glucose and electrolytes
Most preterm infants can’t feed directly from the breast and so milk should be expressed and fed by nasogastric tube during steps 4 – 6.
XXVII. Foetal erythropoiesis and haematology
- This is not a topic on the topic list, but it was discussed in the lectures this semester so it might be good to know
- Place of foetal erythropoiesis
- Weeks 2 – 10: Yolk sac
- Weeks 10 – 26: Liver
- Weeks 18 onwards: Bone marrow
- Foetal erythropoiesis does not depend significantly on the maternal nutritional status
- The foetus, like the parasite it is, is very good at trapping iron, folate, B12 from the mother
- Timing of umbilical cord clamping
- Right after birth, a lot of blood is moved from the placenta to the foetus through the umbilical cord = placental transfusion
- Clamping the umbilical cord too early prevents placental transfusion, lowering the blood volume of the neonate
- However, clamping too late causes polycythaemia
- Haemoglobin structure
- Foetal haemoglobin (α2γ2) has different structure than adult haemoglobin (α2β2)
- Foetal Hb has greater affinity for oxygen, increasing oxygen uptake from maternal blood in the placenta but decreasing peripheral oxygen delivery
- After delivery, synthesis of γ chains decrease while β chains increase, eventually making all Hb molecules in the body α2β2
- Oxygen saturation
- In utero oxygen saturation is 45 – 60% (relative hypoxia compared to adult life)
- After birth, rises to 95%
XXIX. Neonatal anaemia
- Also not a topic on the topic list, but it may be on next semesters list?
- Etiology
- Blood loss
- Abruptio placentae
- Placenta praevia
- Twin-to-twin transfusion syndrome
- Ruptured lived or spleen
- Haemolysis
- Haemolytic disease of the newborn (Rh, ABO incompatibility)
- G6PD deficiency
- Haemoglobinopathies
- Diminished RBC production
- Blackfan-Diamond syndrome
- Infections
- Anaemia of prematurity
- Physiologic anaemia
- Blood loss
- Neonatal blood transfusion
- In severe anaemia blood transfusions are necessary, but these transfusions also cause severe side effects in the neonates and should only be used when absolutely necessary
- For this reason, there are strict guidelines regarding when to transfuse neonates
- Physiologic anaemia
- Normocytic normochromic anaemia which is physiological in neonates
- Pathogenesis
- With the onset of respiration, O2 saturation increases -> EPO production decreases -> RBC production decreases
- During growth after birth, plasma volume increases faster than the RBC mass, causing haemodilution
- Requires no treatment
- Anaemia of prematurity
- = an exaggeration of physiologic anaemia in preterm infants
- Occurs at 3 – 12 weeks after birth in < 32 week preterm infants
- Pathogenesis
- Impaired EPO production for unknown reasons
- Many blood tests taken from preterm infants -> iatrogenic “phlebotomy”
- RBCs of preterm infants have lower lifespan
- Preterm infants grow relatively faster than term infants -> haemodilution
- Preterm infants have more foetal haemoglobin than term infants, so anaemia impairs peripheral oxygenation more in preterms than terms
- Diagnosis
- Hb 60 – 80 g/L
- Clinical features
- Often asymptomatic
- Apnoea
- Tachycardia
- Tachypnoea
- Prevention
- Delay cord clamping
- Reduce number of blood tests taken
- Haemolytic disease of the newborn
- Major problem in the past, nowadays not
- Pathogenesis
- Maternal exposure to foetal antigens on RBCs -> maternal production of antibodies, which cross placenta -> haemolysis of foetal RBCs
- Maternal exposure to foetal RBCs occurs most commonly during birth, so it’s too late for the antibodies to affect the present foetus
- However, if the mother has another foetus with the same RBC antigens, that foetus can be affected
- Incompatibility in ABO antigens -> more common, but mild disease
- Example: Mother O, foetus A
- Incompatibility in Rh antigens -> severe disease, but nowadays prevented
- In Rh- mother with Rh+ foetus
- Clinical features
- Hydrops foetalis
- Only in Rh incompatibility
- Neonatal anaemia
- Jaundice
- Hydrops foetalis
- Diagnosis
- Signs of haemolysis -> Coombs test
- Positive -> Rh incompatibility
- Negative or weak positive -> ABO incompatibility
- Signs of haemolysis -> Coombs test
- Treatment
- Most cases, even Coombs positive ones, don’t require treatment
- Intrauterine blood transfusion
- Only in severe cases
- Phototherapy for hyperbilirubinaemia
- Prevention
- Rh- mothers are screened for anti-D antibodies
- No anti-D antibodies (unsensitized mothers) -> receive anti-D immunoglobulin
- These immunoglobulins will destroy the foetal RBCs during birth before they can sensitize the mother
- Anti-D antibodies present (sensitized mothers) -> monitor foetus for haemolysis
- No anti-D antibodies (unsensitized mothers) -> receive anti-D immunoglobulin
- Rh- mothers are screened for anti-D antibodies
XXX. Haemorrhagic disease of the newborn
- = Vitamin K deficiency bleeding of the newborn
- Pathomechanism
- Neonates have low vitamin K, due to limited transport across placenta, low vit K content in breast milk, and sterile gut
- “Early” type haemorrhagic disease
- Within 24 hours of life
- Due to maternal use of drugs with anti-vitamin K effect
- Warfarin
- Phenytoin
- Phenobarbital
- “Classic” type haemorrhagic disease
- Between day 1 and 7
- Due to exclusive breastfeeding and/or no vitamin K prophylaxis
- “Late” type haemorrhagic disease
- Between 3 weeks and 8 months
- Due to exclusive breastfeeding and/or no vitamin K prophylaxis
- Can also be due to fat malabsorption, AB treatment
- Clinical features
- Bleeding of umbilicus
- Cephalohaematoma
- Intracranial haemorrhage
- Prevention
- All term infants: 2 mg oral vitamin K (Konakion)
- All preterm infants: 1 mg IV vitamin K
- Treatment
- Repeat IV vitamin K
- Fresh frozen plasma transfusion
XXXI. Neural tube defects
- Risk factors
- Maternal folic acid deficiency
- Drugs affecting folate metabolism
- Prevention
- Folic acid before planned pregnancy and during pregnancy
- Myelomeningocele
- = protrusion of both meninges and spinal cord through a defect in the back
- Epidemiology
- 1 – 2 / 1000 live births
- Clinical features
- Paraparesis
- Clubfoot
XXXII. Neonatal hypotonia
- Hypotonia = reduced resistance to passive range of motion in joints
- Sometimes called floppy baby syndrome
- Etiology
- CNS hypoxia/ischaemia
- Genetic syndromes
- Down syndrome
- Prader-Willi syndrome
- Spinal muscle atrophy
- Congenital myasthenia gravis
- Muscular dystrophy
- Metabolic myopathy
- Diagnosis
- Infant’s body forms an upside-down U-shape when held at abdomen or back
XXXIII. Periventricular leukomalacia and hydrocephalus
- Periventricular leukomalacia (= encephalopathy of prematurity)
- Mainly affects premature infants
- The lower the birth weight, the higher risk
- 5% of very low birth weight (< 1500 g) infants
- Pathomechanism
- Hypoxia, ischaemia, or inflammation cause focal necrosis of periventricular white matter
- Clinical features
- Infants rarely show signs, but they appear later
- Typically causes spastic diplegia of the legs
- Can cause weakness of arms and face if severe
- Diagnosis
- Typically diagnosed during routine screening ultrasound
- Mainly affects premature infants
- Hydrocephalus
- = enlarged ventricles due to excess CSF
- 2 types, although a mixture of both types often occurs
- Etiology
- Interventricular haemorrhage
- Infection
- TORCH
- Choroid plexus tumour
- Brain tumour
- Development malformations
- Neural tube defects
- Aqueductal stenosis
- Arnold-Chiari malformation
- Dandy-Walker malformation
- Noncommunicating or obstructive hydrocephalus
- More common than communicating
- Something obstructs the CSF circulation at the point of foramen of Monro, aqueduct of Sylvius, or fourth ventricle
- Communicating hydrocephalus
- Less common than noncommunicating
- Blood or debris obstruct and inflame the arachnoid villi, preventing CSF absorption
- Local inflammation -> inflamed arachnoid villi -> decreased CSF absorption
- Treatment
- Ventriculoperitoneal shunt
- One-way shunt which drains CSF into the peritoneum
- Ventriculoperitoneal shunt
GENERAL PAEDIATRICS
History taking in paediatrics
- Not a topic, but should be known
- Maternal history
- Age
- Chronic diseases
- Number of pregnancies
- Number of children
- Medications
- Problems during pregnancy
- Bleeding
- Trauma
- Hypertension
- Infection
- Drug/alcohol/smoking abuse
- Labour history
- Length of labour
- Type of delivery
- Use of forceps/vacuum
- Use of anaesthesia
- Gestational age (pre-term/mature/post-term)
- Birth weight and length
- APGAR score
- Neonatal history
- Any problems in neonatal period
- Breathing problems
- Use of oxygen
- Need for intensive care
- Need for phototherapy
- Feeding problems
- Feeding history
- Breastfed/bottle fed
- Use of formula
- Frequency, amount
- Should be approx. 150 mL/kg body weight/day
- Other history
- Vaccination history
- Chronic diseases
- Medications
- Allergies
- Family history
- Social history
1. Heart failure
Etiology
- Neonates and infants
- Ventricular septal defect
- Patent ductus arteriosus
- Other CHDs (hypoplastic left heart, aortic valve stenosis, etc.)
- Older children
- Eisenmenger syndrome (untreated acyanotic CHD)
- Rheumatic heart disease
- Any age
- Cardiomyopathy
- Myocarditis
Clinical features
Signs and symptoms of heart failure in children are not much different from those in adults.
- Tachycardia
- Tachypnoea
- Breathlessness
- Hepatomegaly
- Recurrent chest infections
- Poor feeding
- Sweating
- Poor growth
Symptoms usually begin after a few weeks/months of age, as the pulmonary vascular resistance decreases progressively, increasing left-to-right shunting.
Diagnosis
X-ray shows cardiomegaly and increased pulmonary vascular markings, but rarely gives the underlying cause. Echocardiography is essential for diagnosis. For evaluation of cardiomyopathies, MRI may be used.
Treatment
Medical treatment of heart failure is mostly the same as in adults, mostly consisting of diuretics and ACE inhibitor/ARB. Definitive treatment of heart failure involves treatment of the underlying cause, if possible.
2. Infective endocarditis, myocarditis, pericarditis in childhood
Infective endocarditis
Infective endocarditis mostly affects children with structural heart disease, especially congenital heart disease, shunts, VSD patches, etc. Streptococcus viridans accounts for most cases.
Clinical features:
- Fever
- Malaise
- Anaemia, pallor
- Distal emboli causing necrosis in skin, CNS, kidney
- New heart murmur
- Embolic phenomena
- Osler nodes – painful red lesions on hands, fee
- Roth spots – spots on retina
- Janeway lesions – painless lesions on hands, feet
- Splinter haemorrhages – under nails
Diagnosis is made based on positive blood cultures and presence of vegetations on echocardiography. Inflammatory markers are elevated.
Treatment involves empiric antibiotics (penicillin + gentamicin), often given immediately after obtaining blood cultures. Antibiotic treatment lasts for 4 – 8 weeks. If there is infected foreign material in the heart, it may need to be surgically removed.
Prevention is important for those with strutural heart disease. They should practice good oral hygiene and take prophylactic antibiotics before dental and other procedures.
Myocarditis
Myocarditis is due to viral infection, often Coxsackie B or adenovirus. There’s an initial phase of viral infaction characterised by infectious symptoms (fever, myalgia, malaise), followed by a phase of myocardial inflammation. During the latter phase, dilated cardiomypathy and heart failure or arrhythmia can develop.
MRI or endomyocardial biopsy can be used for diagnosis. Viral swabs of rectal and nasal mucosa can be used to confirm viral etiology.
There is no curative therapy. The goal is to stabilize while we wait for it to resolve. We need to treat heart failure if present. Intravenous immunoglobulin (IVIG) may improve prognosis. Physical activity should be limited for 6 months after onset of disease to decrease the risk of complications.
In most cases, myocarditis and dilated cardiomyopathy spontaneously resolve. However, some never fully recover cardiac function, and a few require heart transplant.
Pericarditis
Like myocarditis, pericarditis is often a result of viral infection (enterovirus, influenza. Other causes include uraemia, rheumatoid arthritis, and postpericardiotomy syndrome following heart surgery. The biggest problem of pericarditis is the pericardial effusion which forms secondarily.
Symptoms depend on the size of the pericardial effusion and how fast it accumulates. Small effusions are well tolerated, as a larger ones which have accumulated slowly. A rapidly developing large effusion compromisies the patient’s haemodynamics.
Typical symptoms include pleuritic chest pain (sharp and piercing pain which worsens on deep inspiration or when lying down), fever, and symptoms of pericardial effusion like dyspnoea, distended neck veins.
Auscultation reveals distant heart sounds and a pleural friction rub. X-ray shows cardiomegaly with a roundish heart. ECG shows widespread ST elevation and reduced voltage. Echocardiography shows the pericardial effusion.
In case of large symptomatic pericardial effusions, pericardiocentesis may be required. There is no other treatment for viral pericarditis than NSAIDs.
3. Otitis, sinusitis
Acute (suppurative) otitis media
Infants and young children are predisposed to acute otitis media (AOM) due to having short, horisontal Eustachian tubes. Every febrile child should be examined for AOM.
Symptoms include ear pain, fever, and possibly hearing loss. Otorrhoea may develop if there is a perforation. Complications like mastoiditis and meningitis are rare.
Physical examination of the tympanic membrane with an otoscope shows a white/yellow and bulging membrane with loss of the normal light reflection. The diagnosis is made based on these physical examination findings.
Treatment is analgesics (paracetamol/NSAIDs) empiric antibiotics, usually amoxicillin or amoxicillin-clavulanic acid. In low-risk children (> 2 years, unilateral AOM, not severe symptoms) we may opt for observation rather than antibiotics. AOM usually resolves spontaneously, but antibiotics may shorten the duration of symptoms.
If the child appears toxic or antibiotics fails to improve the condition, tympanocentesis and bacterial culture of the fluid should be taken.
Otitis media with effusion
Otitis media with effusion (OME), also called glue ear, is characterised by middle ear effusion without infection. It may develop after acute otitis media. It’s a very common condition which affects almost 90% of children by the age of 4.
OME is mostly asymptomatic, but it may cause hearing loss and tinnitus. Physical examination with otoscope shows bubbles or air-fluid level behind the tympanic membrane without signs of infection (bulging, fever, pain).
In most cases, OME spontaneously resolves. If it doesnt, a tympanostomy tube (grommet) may be placed to allow for drainage for some months. Adenoidcteomy may also help.
Otitis externa (= swimmer’s ear)
Otitis externa, also called swimmer’s ear, refers to inflammation of the external auditory canal due to bacterial infection. Like the nickname suggests, it’s associated with the entrance of bacteria into the ear during swimming. Pseudomonas and staph are the most common causes.
Symptoms include severe ear pain, especially at night, itching, and otorrhoea. Physical examination with otoscope shows signs of infection in the external auditory meatus (erythema, swelling). The diagnosis is made clinically.
Treatment is antibiotic eardrops with or without topical glucocorticoids. Systemic antibiotics may be used for severe cases.
Rhinosinusitis
Infection of the nasal cavity and paranasal sinuses is often part of a regular viral upper respiratory tract infection (URI), and is called viral rhinosinusitis. This causes symptoms like nasal discharge, cough, and fever (usually mild). Typically symptoms peak in severity on days 3 – 6 and then improve progressively.
Rarely, a secondary bacterial infection may develop as a result of obstruction of mucuous flow from the mucosal oedema from a URI. In these cases, pain, swelling, and tenderness over the cheek develops from infection of the maxillary sinus. The frontal sinuses don’t develop until late childhood, so frontal sinusitis is uncommon in young children.
Bacterial rhinosinusitis symptoms persist for more than 10 days without improving. Worsening of rhinosinusitis symptoms may be a sign that secondary bacterial rhinosinusitis has developed. The child may also appear ill, have higher fever (> 39°C), have purulent nasal discharge, and severe headache. “Double sickening”, the phenomenon where symptoms initially improve before they worsen, may also be present.
Haemophilus influenzae, pneumococcus, and moraxella are the most common causes of bacterial infection. Amoxicillin-clavulanic acid is used for treatment. If the patient is in a severe condition, they should be hospitalised.
4. Epiglottitis, laryngitis
Epiglottitis
Epiglottitis is a medical emergency, as the airway can suddenly become obstructed. Tha majority of cases are due to Haemophilus influenzae type b, and so the introduction of the Hib vaccine in many countries’ childhood vaccination programme has made epiglottitis very rare in these countries. It may still occur due to other bacteria, but is very rare overall.
The patient presents as very ill-looking, with high fever and drooling saliva (as they cannot swallow). Symptoms develop over the course of hours rather than days. There is inspiratory stridor, and the patient may assume a “sniffing” posture with their head forward and mouth open.
If epiglottitis is suspected, the patient should be admitted to the hospital urgently. Protecting the airway is the first priority, but laryngoscopy and intubation can only be performed under controlled conditions in an operating room and under general anaesthesia, as any manipulation of the airways may trigged airway obstruction.
After the airway is secured, blood culture and epiglottic culture is taken to determine the causative bacterium. Afterward, empiric antibiotics are initiated.
Croup
Croup (acute laryngotracheobronchitis) refers to viral infection of the larynx, trachea, and bronchi. It’s a very common condition which occurs between the age of 6 months – 6 years, but incidence peaks in 2-year olds. It is most common in the autumn.
It’s most commonly caused by parainfluenza virus, but it may also be caused by rhinovirus, RSV, and influenza.
Croup is usually preceded by a common cold (URTI). The typical features of croup are:
- Inspiratory stridor
- Hoarseness, wheezing
- Barking seal-like cough
- Difficulty breathing with chest recession
Symptoms are usually mild, but worsen at night. The diagnosis is based on clinical features. X-ray may show subglottic narrowing.
Most cases are mild and can be managed at home, but the parents must be educated on signs of severe airway obstruction. Croup is self-limiting. It’s important to calm the child, as anxiety worsens the airway obstruction. Nebulised epinephrine inhalation can be used to rapidly (but transiently) improve the airway obstruction. Inhalation of cool, moist air is often used (but not evidence-based).
Glucocorticoids reduce the severity and duration of croup. They can be administered orally or IM (dexa, prednisolone) or by inhalation (nebulised budesonide).
(Bacterial tracheitis)
Bacterial tracheitis is caused by staph aureus, usually after a viral upper respiratory tract infection. The clinical picture is very similar to those of croup and epiglottitis, with ill appearance, stridor, cough, and respiratory distress. If a patient with croup-like symptoms is treated like croup (nebulised epinephrine) but the condition is not improving, one should consider bacterial tracheitis. A culture is made from samples from the trachea obtained during bronchoscopy, after which empiric IV antibiotics are initiated.
5. Pharyngitis, tonsillitis
Pharyngitis is most commonly viral (2/3 of cases), but is most severe when bacterial. Bacterial pharyngitis is caused by group A streptococci (GAS), often callsed “strep throat”. Tonsillitis is characterised by purulent exudate on the tonsils, and may be present in GAS pharyngitis and mononucleosis.
Symptoms of streptococcal pharyngitis include sore throat, fever, and dysphagia. Cervical lymphadenitis is common. Physical examination of the throat shows an erythematous, oedematous pharynx and tonsils. Exudates on the tonsils are especially characteristic.
A POCT antigen test for streptococci exists and can be used to confirm the diagnosis, but a throat culture is an alternative. The so-called Centor score can be used to estimate the likelihood of GAS pharyngitis. If the likelihood is low, the antigen test is unnecessary.
Treatment is with antibiotics. The antibiotic of choice is penicillin. Amoxicillin should be avoided, as it can cause a rash if mononucleosis has been misdiagnosed as GAS pharyngitis. GAS pharyngitis is self-limiting, and antibiotics don’t significantly shorten the disease duration, but they prevent complications such as rheumatic fever and poststreptococcal glomerulonephritis. If a patient develops recurrent, airway-obstructing tonsillitis, tonsillectomy may be necessary.
Streptococcal pharyngitis may present with scarlet fever, a delayed-type hypersensitivity reaction to the GAS toxin. In this case, the patient develops not only streptococcal pharyngitis and tonsilliis but a strawberry red tongue and a diffuse erythematous maculopapular rash which gives a “sandpaper” quality to the skin. Treatment is the same.
6. The seriously ill child (recognition, evaluation, monitoring)
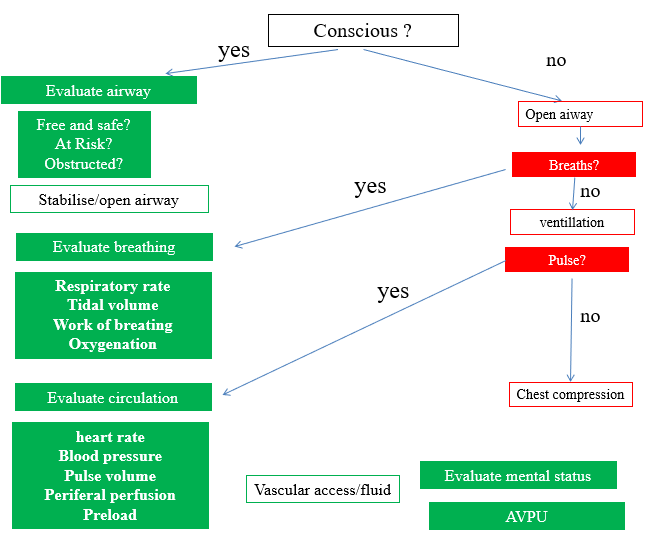
A child who is unconscious or has symptoms like respiratory distress may be seriously ill and require urgent stabilisation. If this interaction occurs in the environment, it’s important to ensure your own safety and the safety of the child before proceeding. Call for help as soon as possible.
Evaluate responsiveness
Attempt to establish contact by speaking loudly to the child and attempting painful manoeuvres like pinching, gently shaking, or pressing on the fingernails.
If the child is responsive, proceed to evaluating the airway. If the child is unresponsive, proceed to opening the airway.
A – airway
Unconscious children obstruct their airway and need airway-opening menoeuvres or airways adjuncts to hold their airway open. A person who is conscious and talking has a patent airway. Signs of an already or imminently compromised airway in a conscious person include being unable to talk, stridor, cyanosis, and respiratory distress. If an obstructing foreign body can be visualised, attempt to remove it.
Perform the head tilt-chin lift manoeuvre by tilting the head back and lift the chin. Don’t tilt the head of infants (< 1 year) back, but perform chin lift (their neck should remain in a neutral position).
If neck injury is suspected, don’t perform the head tilt-chin lift manoeuvre but rather the jaw thrust manoeuvre, in which you grasp the angles of the lower jaw and move the mandible forward.
After attempting a manoeuvre, evaluate breathing. If these manoeuvres fail to restore airway patency, we may place an airway adjunct like an oropharyngeal or nasopharyngeal airway. There are especially indicated if it’s suspected that oropharyngeal structures like the tongue or hypertrophic tonsils are obstructing the airway.
If the child is conscious but with a suspected foreign body obstruction, perform:
- 5 thrusts against the back followed by 5 thrusts against the chest for infants (< 1 year)
- 5 thrusts against the back followed by 5 thrusts against the abdomen (Heimlich) for older children (> 1 year)
B – breathing
While maintaining the airway, evaluate breathing by placing your ear over the child’s mouth. Listen and feel for breathing, while observing the chest for air movement. Spend no more than 10 seconds on this step. If there is abnormal or no breathing, give 5 rescue breaths while evaluating for whether the chest is elevating; if not, the airway is not patent. When administering breaths, either by mouth-to-mouth, mouth-to-nose (in infants), or by bag-mask ventilation, only inflate the lungs until you see the chest rise. Don’t hyperinflate the lungs.
Check for signs of life after these rescue breaths. If the person is spontaneously breathing, place them in the recovery position. If not, continue to C.
C – circulation
Feel for a pulse. In an infant, check the brachial or femoral pulses. In an older child, check the carotid or femoral. If there’s no pulse, or the heart rate is < 60 bpm and there are signs of poor circulation, initiate chest compressions.
Give 15 chest compressions, followed by 2 breaths, and repeat. Compressions should be at a rate of 100/minute and administered to the lower half of the sternum. Effective chest compressions require a compression depth of 1/3 of the depth of the thorax, with complete recoil after each compression.
Monitoring
Critically ill children are usually treated in the paediatric/neonatal ICU, and continuously monitored using the following:
- Clinical signs
- Capillary refill time – normal is <3 seconds
- Colour of extremities – look for cyanosis/pallor
- Temperature of extremities – look for cold extremities
- Urine output – patients are catheterised, making monitoring output easy
- State of consciousness – assessed by AVPU or GCS
- Core temperature – rectal or inside bladder catheter
- Non-invasive monitoring
- Heart rate
- Respiratory rate – calculated by ECG electrodes
- Pulse oximetry (SpO2) – normal is > 95%
- ECG – simple monitoring (only leads I – III) or 12-lead
- End-tidal CO2 concentration (ETCO2) – normal is 2 – 3 mmHg less than PaCO2
- Echocardiography – assesses cardiac function and fluid responsiveness
- Invasive monitoring
- Blood test or arterial blood gas
- Arterial blood pressure – invasive or non-invasive
- Central venous pressure (CVP) – assesses intravascular volume and cardiac function
- Central venous oxygen saturation (ScvO2)
- PiCCO – uses transpulmonary thermodilution to provide information on cardiac output, fluid status, +++
- Intracranial pressure monitoring – indicated for GCS 8 or less, or for severe head trauma
Normal values in paediatrics:
| Pulse | Systolic BP | Respiratory rate | |
|---|---|---|---|
| Neonate | 100 – 160 | 50 – 70 | 35 – 60 |
| Infant | 80 – 140 | 80 – 100 | 30 – 40 |
| Young child | 80 – 110 | 90 – 110 | 20 – 30 |
| School-age child | 70 – 100 | 100 – 120 | 20 – 25 |
| Adolescent | 60 – 90 | 110 – 130 | 15 – 20 |
7. Assessment of nutritional status (percentile, BMI, skin fold, laboratory tests). Forms of malnutrition
Assessment of nutritional status
When assessing the nutritional status and growth of a child, we must use percentile charts. These charts compare the parameters of the child to those of other children of the same sex, age, and height. Percentile charts exist for weight, BMI, waist circumference, hip circumference, etc.
For example, a child in the 95th percentile for weight is heavier than 95% of children of the same sex, age, and height. A child in the 50th percentile (the median) for weight is heavier than 50% of the children of the same sex, age, and height, i.e. they’re right in the middle.
It’s important to use local percentile charts as these characteristics are different from area to area.
In children, BMI must be assessed as a percentile. A child is overweight if their BMI is in the 85th – 95th percentile, and obese if it’s in the 95th percentile or higher. A skinfold calliper is a cheap method of evaluating the distribution and amount of fat. It can be used on the triceps or abdomen.
Some laboratory parameters can be used to assess the nutritional status:
- Microcytic anaemia -> likely iron deficiency
- Macrocytic anaemia -> likely folate, B12 deficiency
- Prealbumin = marker of short-term dietary adequacy
- Albumin = marker of long-term dietary adequacy
- Serum vitamin D = marker of vitamin D intake
- Ferritin, transferrin, serum iron = markers of iron intake
Other laboratory tests may be useful in evaluation of comorbidities of obesity, like HbA1c, lipid panel, thyroid function, liver function, etc.
Malnutrition and failure to thrive
Malnutrition refers to deficiencies, excesses, or imbalances in a person’s intake of energy and/or nutrients. It can lead to:
- undernutrition, which includes wasting (low weight-for-height), stunting (low height-for-age) and underweight (low weight-for-age);
- micronutrient-related malnutrition, which includes micronutrient deficiencies (a lack of important vitamins and minerals) or micronutrient excess; and
- overweight, obesity and diet-related noncommunicable diseases (such as heart disease, stroke, diabetes and some cancers).
The situation where there is sub-optimal weight gain or rate of weight gain in young children over time is called failure to thrive or faltering growth. There are no universally accepted definitions for failure to thrive. Commonly used definitions include:
- Weight below 2nd percentile for age
- Weight decreasing, crossing two major percentile lines on the growth chart over time
- Head circumference below 2nd percentile for age
This is evaluated based on percentile charts plotting weight for age, weight for height, and height for age.
The severity of malnutrition can be graded according to the Waterlow classification:
- No malnutrition – weight > 90% of expected weight for age
- Mild malnutrition – weight 80 – 90% of expected weight for age
- Moderate malnutrition – weight 70 – 80% of expected weight for age
- Severe malnutrition – weight < 70% of expected weight for age
Feeding of malnourished infant is a long process which takes months. In severe cases, it should be performed in-hospital. It should start slowly to prevent refeeding syndrome. Initially, the diet should contain twice as much energy, protein, and lipid as the recommended daily. The child should be treated with simultaneous psychotherapy. Over time, the weight gain increases back to baseline.
Some children grow slowly physiologically. Here are some features with which we can differentiate constitutional slow growth from faltering growth.
| Slow growth | Faltering growth | |
| Weight gain | Continuous | Not continuous |
| Feeding | Normal frequency | Less frequent |
| Muscle tone | Good | Hypotonic |
| Skin turgor | Good | Decreased |
| Behaviour | Normal | Abnormal |
| Urination | Normal | Less frequent |
| Defecation | Normal | Less frequent |
8. Vitamin deficiencies (fat- and water-soluble vitamins), trace elements and their deficiencies
Iron deficiency
Term infants don’t need iron supplement until 4 months of age. Preterm infants have much smaller iron stores and higher iron needs, and so they need iron supplement with breast milk or iron-fortified formula. The iron requirements in infancy are:
- Term infants: 1 mg/kg of body weight/day
- Preterm infants: 2 mg/kg of body weight/day
Too early introduction of cow’s milk (whose iron has low bioavailability) can lead to iron deficiency. Cow’s milk should not be introduced (at least in large amounts) until 12 months of age.
Clinical features of iron deficiency include microcytic anaemia, spoon nails, reduced muscle and mental performance, and pica.
Vitamin C deficiency
Vitamin C deficiency causes scurvy, but is rare in children. It may occur if they’re fed a diet containing no fruits and vegetables. Scurvy is characterised by petechiae, bruising, bleeding gums, and painful joints.
Vitamin B1 (thiamine) deficiency
Thiamine deficiency causes beri-beri, but is rare in children. It may occur in children breastfed by alcoholic mothers, alcoholics, or in those who boil milk, as boiling destroys the vitamin. Beri-beri is characterised by polyneuropathy and heart failure. Infants with beri-beri have a characteristic aphonic cry, where they appear to be crying but there’s no sound.
Vitamin B3 (niacin) deficiency
Tryptophan can be converted to niacin, so deficiency only occurs if both tryptophan and niacin in the diet are deficient. This is rare.
Niacin deficiency causes pellagra, which is characterised by the 4 D’s:
- Dermatitis
- Diarrhoea
- Dysphagia
- Dementia
Vitamin B9 (folate) deficiency
Folate deficiency can develop within a few weeks of birth as newborns have 10x the folate requirement as adults (relative to body weight), but they have no stores of folate. This vitamin is heat labile, so heating formula too much decreaes the folate content.
Folate deficiency in utero causes neural tube defects. During life, folate deficiency causes megaloblastic anaemia.
Vitamin B12 deficiency
B12 deficiency is rare in childhood. If it occurs, it’s due to pernicious anaemia rather than malnutrition. Deficiency causes megaloblastic anaemia, polyneuropathy, and neurological signs.
Vitamin A deficiency
Vitamin A deficiency is mostly a disorder of those with fat malabsorption (like cystic fibrosis) and children in developing countries who don’t receive supplement. Hypovitaminosis A causes xerophthalmia, characterised by dry eyes and night blindness, as well as increased susceptibility to infection.
Vitamin D deficiency
Vitamin D deficiency is a relatively common disorder of childhood. In most cases, it’s due to inadequate sunlight (UVB) exposure rather than dietary deficiency. Hypovitaminosis D in children causes hypocalcaemia and hypophosphataemia, eventually developing rickets, which is characterised by:
- Craniotabes – Thinning of the skull
- Rachitic rosary – enlargement of costochondral junction
- Thickening of wrists and ankles
- Bowlegs
Vitamin K deficiency
Neonates are naturally vitamin K deficient, which is why they’re routinely administered a vitamin K shot (Konakion®) i.m. after birth. The dose is 1 mg. This prevents haemorrhagic disease of the newborn, characterised by generalised ecchymoses, GI bleeding, and intracranial haemorrhage.
9. Congenital and acquired immunodeficiencies
Congenital immunodeficiency
Congenital (primary) immunodeficiencies are rare conditions, and there are more than 250 recognised disorders. Most are due to single gene defects.
Congenital immunodeficiencies may present in different manners, but they should be suspected in children with SPUR infections. SPUR is an acronym which stands for:
- Severe (like meningitis, peritonsillar abscess)
- Persistent (don’t improve with usual antibiotics)
- Unusual (pathogens, like PCP or Burkholderia)
- Recurrent
Chronic diarrhoea and poor growth are other typical features of congenital immunodeficiencies. There’s often also an increased risk of autoimmune disease and cancer.
We can classify the congenital immunodeficiencies according to which part of the immune system which is defective, each of which have typical clinical features.
B-cell (antibody) defects
B-cell defects are the most common congenital immunodeficiencies. They present after month 4 (as they’re protected by the maternal antibodies during this time), but during the first 2 years.
B-cell defects predispose to bacterial infections, especially infections by encapsulated bacteria. The patient develops recurrent severe respiratory infections, which can lead to bronchiectasis.
These are the most important types of B-cell deficiency:
- Selective IgA deficiency – quite common (1:500), but it’s often asymptomatic and therefore unnoticed. It may predispose to respiratory infections.
- X-linked (Bruton) agammaglobulinaemia – characterised by absence of antibodies. Characteristically, there is no lymphoid tissue, so no lymph nodes or tonsils.
- Common variable immune deficiency (CVID) – presents in the 20s/30s.
- Hyper IgM syndrome is characterised by defective B-cell isotype switching, causing B-cells to only be able to produce IgM antibodies.
- Transient hypogammaglobulinaemia of infancy – a condition characterised by abnormally low antibody levels in infancy, but the levels normalise after a few years.
On the exam, I was asked about which condition which would be difficult to diagnose in someone with selective IgA deficiency. The answer is Coeliac disease, because in this disease detection of IgA antibodies is important.
To evaluate for B-cell defects, it’s important to measure the number of B-cells and the level of each of the antibody classes.
B-cell defects are often given lifelong antibotic prophylaxis to prevent recurrent bacterial infections. They may also receive immunoglobulin replacement therapy.
T-cell and combined immunodeficiencies
T-cell defects often causes features of B-cells defects due to how T-cells activate B-cells, so the distinction between T-cell deficiency and combined deficiency is not sharp. These defects increase the risk for viral and fungal infection. They present in the first months of life.
These are the most common types of T-cell defects and combined immunodeficiencies:
- Severe combined immunodeficiency (SCID) – a heterogenous group of disorders with profound both T-cell and B-cell defects.
- Wiskott-Aldrich syndrome – an X-linked disorder characterised by the triad of immunodeficiency, thrombocytopaenia, and eczema.
- DiGeorge syndrome – a genetic syndrome caused by deletion of chromosome 22q11.2, which causes abnormal development of the third and fourth pharyngeal pouches, causing abnormal thymus, parathyroid, and face. It’s characterised by cardiac defects, palatal defect, abnormal face, absent thymus, and hypocalcaemia.
- Ataxia telangiectasia – a disorder characterised by a defect in DNA repair. Like the name suggests, it causes cerebellar ataxia and telangiectasias in addition to immunodeficiency.
- X-linked lymphoproliferative disease – characterised by inability to generate a normal response to Epstein-Barr virus. The patient either dies from the infection or develops secondary lymphoma after clearing it.
To evaluate for T-cell defects, it’s important to measure the number of each T-cell type. If a combined immunodeficiency is suspected, also evaluate as for a B-cell defect.
Bone marrow transplantation is an option for certain T-cell disorders, like SCID. Lifelong prophylaxis with TMP-SMX for PCP and antifungals is commonly used.
Neutrophil defects
Neutrophil defects are rare. They’re characterised by recurrent bacterial infections, abscesses, poor wound healing, and granulomas. The most common type is chronic granulomatous disease.
Acquired (secondary) immunodeficiency
Acquired (secondary) immunodeficiency is more common than primary. It can be caused by:
- HIV
- Hyposplenism/splenectomy
- Immunosuppressant drugs
- Antibody loss (nephrotic syndrome, burns)
- Diabetes mellitus
- Malnutrition
- Malignancy
10. Juvenile idiopathic arthritis
Juvenile idiopathic artritis (JIA) is the most common chronic inflammatory rheumatic disorder or children. It’s defined as the presence of arthritis for more than 6 weeks presenting before 16 years of age, after other etiologies have been excluded. It affects 1:1000 children and is 5x more common in females.
There are five major types:
- Oligoarthritis – affects ≤ 4 joints
- Polyarthtitis – affects more than 4 joints
- Systemic JIA (Still disease) – also has fever and rash
- Psoriatic arthritis – also has psoriasis
- Enthesitis
Typical clinical features are swelling, stiffness after periods of rest, morning joint stiffness, and pain. Erythema is absent and would rather indicate septic arthitis. Painless chronic uveitis is a possible complication which can lead to vision loss.
The diagnosis is made based on clinical features after other etiologies have been excluded, including early-onset rheumatoid arthritis, septic arthitis, reactive arthritis, etc. There may be leukocytosis, elevated ESR, and elevated CRP. ANA and RF may be positive, both of which are negative prognostic factors. After diagnosis the patient should be screened regularly by an ophthalmologist for uveitis.
NSAIDs are used for symptomatic treatment. Intraarticular glucocorticoids, methotrexate, and biological therapy reduce joint damage. Early aggressive treatment improve outcomes and prevent complications like joint damage and contractures.
11. Systemic autoimmune diseases (SLE, antiphospholipid syndrome)
Systemic lupus erythematosus
Childhood SLE (cSLE) or juvenile-onset SLE (JSLE) is the same disease as in adults, but usually more severe. It mostly affect female teenagers, and Asian and Black ethnicities more often than others.
Most common clinical features are malaise, arthralgia, photosensitivity, and butterfly rash, although other symptoms like fatigue, fever, mouth ulcers, lymphadenopathy etc., may also be present. Symptoms appear and worsen over weeks to months. Organ involvement (serositis, renal disease, seizures or psychosis) are serious complications.
The diagnosis of cSLE is the same as in adults. We may use the classic “4 out of 11” ACR criteria or the newer (more complicated but more sensitive and specific) EULAR/ACR criteria. The following laboratory parameters are typical:
- Positive ANA (must be present for diagnosis)
- Positive anti-dsDNA
- Leukopaenia
- Thrombocytopaenia
Treatment involves DMARDs like hydroxychloroquine, glucocorticoids, cyclophsphamide, etc. NSAIDs are used for symptomatic treatment of musculoskeletal manifestations.
Antiphospholipid syndrome
Antiphospholipid syndrome is characterised by an increased predisposition to thrombosis. It may be primary (idiopathic) or secondary to SLE or other autoimmune diseases.
Typical manifestations include DVT, cerebral venous sinus thrombosis, and ischaemic stroke. Antiphospholipid antibodies like anticardiolipin may be present in the serum. Treatment is by antithrombic prophylaxis with aspirin with or without warfarin/LMWH.
12. Juvenile dermatomyositis
Juvenile dermatomyositis is very rare, affecting only 2 – 4 per million children. It’s characterised by symmetric proximal muscle weakness, heliotrope rash (pink-purple rash on the upper eyelids), and periorbital oedema. Symptoms like fever and fatigue are also common.
CK is usually elevated, and inflammatory markers may be as well. Muscle biopsy shows inflammatory cell infiltrate and atrophy. MRI and EMG show features of myositis.
Treatment is physiotherapy and immunosuppressants like glucocorticoids, methotrexate, etc.
13. Kawasaki disease, Henoch-Schönlein purpura
Kawasaki disease
Kawasaki disease is a form of systemic vasculitis. It mostly affects children of Japanese ethnicity in the age of 1 – 5.
There are no diagnostic tests for Kawasaki disease. It is diagnosed when the child has had fever for at least 5 days and four out of these five criteria:
- Bilateral nonexudative conjunctivitis
- Changes in the oral mucosa (red cracked lips, inflamed mucosa, strawberry tongue)
- Cervical lymphadenopathy (usually unilateral)
- Polymorphic rash
- Erythema and oedema on the hands and feet or desquamation around the nails
Inflammatory markers are elevated. Differential diagnoses like scarlet fever, toxic shock syndrome, and Stevens-Johnson syndrome must be excluded.
The major complication of Kawasaki disease is coronary artery aneurysm, which can lead to myocardial ischaemia. It’s important to recognise and treat the disorder early to reduce the risk of complications. Echocardiography should be performed at diagnosis and after 1 month to look for aneurysms.
Treatment is IVIG and aspirin.
Henoch-Schönlein purpura (HSP)
Henoch-Schönlein purpura (HSP), also called IgA vasculitis, is another form of vasculitis. HSP mostly occurs in the winter, and typically in boys of the age 3 – 10.
It occurs due to deposition of IgA immunocomplexes in vessel walls. It usually occurs 1 – 3 weeks after an upper respiratory tract infection, either by GAS or viruses.
Clinical features include:
- Purpuric rash (on buttocks and extensor surfaces, spares the trunk)
- Arthritis/arthralgia
- Colicky abdominal pain
- Renal symptoms (haematuria/proteinuria/nephritic syndrome)
The diagnosis is clinical, based on typical clinical features, especially the characteristic rash. However, it’s notable that there is thrombocytosis, as most conditions characterised by purpura is associated with thrombocytopaenia.
HSP is self-limiting, and most cases are mild and only need supportive treatment. Those with severe symptoms require systemic glucocorticoid therapy.
14. The milestones of the normal psychomotor development
- Most important developmental milestones
- In the lecture prof. Hollódy said we shouldn’t know the specific milestones at specific ages, but rather the biggest milestones like:
- At 4 months they should be able to turn themselves
- At 5 months they should be able to sit
- At 12 months they should be able to walk
- In the lecture prof. Hollódy said we shouldn’t know the specific milestones at specific ages, but rather the biggest milestones like:
Gross motor milestones:
| Mean age (months) | Milestone |
|---|---|
| 1 | Head up in prone position |
| 2 | Chest up in prone position |
| 3 | Up on elbows in prone position Head control |
| 4 | Up on hands in prone position Roll over to supine position |
| 5 | Sit supported (tripod position) |
| 8 | Crawl |
| 12 | Walk |
| 15 | Run |
Fine motor milestones:
| Mean age (months) | Milestone |
|---|---|
| At birth | Fisted |
| 3 | Unfisted |
| 4 | Reach and grab |
| 6 | Rake from the surface |
| 9 | Starting to pincer grasp |
| 12 | Voluntary release |
Thera are also milestones related to hearing, speech, and language, as well as social, emotion, and behaviour.
15. Evaluation of the child with developmental delay
Introduction
Impaired/delayed psychomotor development refers to when a child <5 years has not met excepted developmental milestones. If milestones in multiple areas of functioning haven’t been met, the condition is called global developmental delay (GDD). In a way, GDD is the counterpart of intellectual disability in children <5.
Intellectual disability (ID, old name: mental retardation) is characterised by deficits in intellect and functioning, which in general can only be assessed after the age of ~5 years. If a child <5 years has impairment in intellect and functioning which causes them to not meet expected developmental milestones, the appropriate term is global developmental delay.
The prevalence of ID is about 1%, but most who are affected only have mild disability. Most children with intellectual disability present before the age of 5 because a parent is worried about developmental delay.
Severity of intellectual disability is often classified according to IQ, but formal IQ testing is rarely performed in children with suspected ID:
- Mild – IQ 50-70
- Moderate – IQ 35-50
- Severe – IQ 20-35
- Profound – IQ below 20
When a child presents with possible intellectual disability, it’s important identify it early in order to initiate proper management as early as possible.
Etiology of ID and GDD
- Idiopathic (30%)
- Prenatal causes
- Chromosomal disorders (Down, Fragile X)
- Genetic disorders (Rett, Prader-Willi)
- Foetal alcohol syndrome
- Congenital infection (TORCH)
- Teratogens (valproate, lead)
- Inborn error of metabolism
- Perinatal events (asphyxia, trauma, infection, intracranial haemorrhage)
- Postnatal/acquired events (trauma, intracranial haemorrhage or infection, hypothyroidism, CNS neoplasm)
In addition to this, there are multiple conditions which are associated with intellectual disability:
- Autism spectrum disorder
- ADHD
- Depression
- Cerebral palsy
- Hearing loss
- Seizures
- Abuse
Indications for evaluation
Evaluation of the child’s psychomotor development is indicated when it’s suspected (by parents or provider) that the child has a developmental delay.
Here are some examples of findings which may suggest developmental delay:
| Age | Finding |
| Birth and at any age | Lack of response to sound
Lack of interest in interacting with people |
| 4 months | Lack of any drive to communicate
Child does not socially smile |
| 6 – 9 months | Child does not coo or babble
Child not interested in peek-a-boo Child does not laugh in playful situation Poor sound localization Lack of responsiveness to sound |
| 12 months | No verbal routines
Failure to say mama or dada Child does not search for dropped objects |
| 15 – 18 months | No single words
Poor understanding of language |
| 2 years | Knows < 50 words
No phrases with two words Less than 50% of speech is intelligible to strangers Child avoids eye contact Child kicks, bites, screams without provocation |
| 3 years | Frequent repetition of others’ speech
Flat or stilted intonation Less than 25% of speech is intelligible to strangers |
- Indications for evaluation of language, speech, and hearing
- Family history of deafness
- Prematurity
- Congenital anomalies
- Intrauterine infection
- Delayed language development
- Parental concern about hearing loss (should always be taken seriously)
- Indications for visual evaluation
- Intrauterine infection
- Prematurity
- Cerebral palsy
- Strabismus
- Nystagmus
- Cataract
Evaluation
A detailed history about prenatal, perinatal, and postnatal events is important. A three-generation family pedigree should be obtained. These may give information which suggests an etiology. Affection of only males could point to an X-linked etiology.
Physical examination should involve measurement of body parameters (weight, height, head circumference), examination for skin lesions which may suggest genetic disease, complete neurological examination, observation of the child’s behaviour, activity, communication, and mood, as well as interactions between parent(s) and child. Some physical examination findings can suggest ceratain etiologies. The child should also undergo vision and hearing screening.
It’s important to detect features like:
- Micro/macrocephaly
- Abnormal stature
- Short -> Turner syndrome
- Tall -> Sotos syndrome
- Dysmorphic features
- Skin abnormalities
- Caf´e au lait spots -> neurofibromatosis
- Hypomelanic macules, facial angiofibromas -> tuberous sclerosis
- Neurological symptoms (gait, weakness, spasticity, tone)
- Eye abnormalities
- Organomegaly -> metabolic/storage disorders
- Obesity -> Prader-Willi/Beckwith-Wiedemann syndrome
- Abnormal genitalia
- Macroorchidism -> Fragile X syndrome
- Hypogonadism -> Prader-Wili syndrome
It’s important to use percentile curves, which show how the percentage of children who have reached a specific milestone at a specific age. As an example:
- 10% of children have said their first word by month 9
- The average age at which a child says their first word is around month 12
- 90% of children have said their first word by month 14
- If a child hasn’t said their first word by month 14, its language development is in the 10th percentile
Screening tests like Bayley scoring system or Brunet-Lézine scale can be used to screen for developmental delay, while other tools (like Wechsler’s tests) can be used to assess the degree of disability. The Bayley scoring systems are used to assess infant development in different domains. It gives a developmental quotient (DQ), the normal value of which is 100. Other tests which may be indicated are genetic tests, neuroimaging, metabolic work-up, evoked potentials, and EEG.
Clinical features of autism in children (not sure if important)
- Delayed developmental milestones in language
- Unusual speech
- Echolalia (repeating others’ speech)
- Grunting, squealing
- Speaks only about specific topics of interest
- Prefers to use single words rather than longer sentences
- Reduced or absent nonverbal communication
- Reduced or absent response to others’ gestures or facial expression
- Reduced or absent response when called by name
- Lack of social smile or eye contact
- Lack of interest in other children
- Lack of awareness of other people; appearing to be in their own world
- Prefers to play alone
- Repetitive motor mannerisms
- Resistance to change, insisting on following same routines
- Atypical play
- Likes to line up objects, etc.
- Becoming upset if others interfere with their games
16. Diabetes mellitus
Diabetes mellitus type 1
Diabetes mellitus type 1 (T1D) is the most common form of diabetes mellitus in childhood. It’s caused by destruction of pancreatic β-cells by an idiopathic autoimmune process. There’s a genetic predisposition and an association with other autoimmune disorders.
Hyperglycaemia occurs when 80 – 90% of the β-cell mass is destroyed. The typical clinical features are polydipsia, polyuria, and weight loss. There may be a compensatory polyphagia due to the calorie loss. Patients may also present with diabetic ketoacidosis.
The diagnosis of diabetes mellitus is made based on either of these findings:
- Fasting glucose > 7 mM
- Random glucose > 11,1 mM
- OGTT > 11,1 mM
- HbA1c > 6,5% (48 mmol/mol)
In most cases, clinical features are sufficient to differentiate T1D from T2D. Autoantibodies and decreased C-peptide levels are features of T1D and can be used to help differentiate them. The most common autoantibodies are anti-GAD, IA-2 antibodies, and islet cell antibodies.
Long-term hyperglycaemia leads to severe complications, and so the aim of treatment is to maintain normoglycaemia as much as possible, while avoiding hypoglycaemia. A good target is HbA1c < 7,5% (59 mmol/mol).
Lifelong insulin injection therapy is required. The insulin regimen which is most commonly used is a combination of fast-acting insulin injections with meals and long-acting “basal” insulin injection at bedtime. The dose of the fast-acting insulin should be adjusted to the calorie intake. The total carbohydrate content of the foods should be relatively constant and balanced to prevent hypo- and hyperglycaemia. Patients should monitor their blood glucose regularly, especially before meals.
An insulin pump is a device which continuously administer small doses or insulin subcutaneously, and which can administer additional boluses for each meal.
The “honeymoon period” is a phenomenon where patients with recently diagnosed T1D who just started treatment, β-cell function can temporarily recover slightly, reducing the need for insulin. It begins in the first weeks of therapy, and may last for months. After this period, the insulin requirement increases.
Hypoglycaemia occurs when an inappropriate insulin dose is administered relative to the carbohydrate intake. This occurs 1 or 2x a week even in well-controlled T1D, and 10% of patients have a severe episode every year. It should be treated by glucose tablets or sugary drinks.
The late complications of DM are retinopathy, cardiovascular disease, cerebrovascular disease, nephropathy, etc. Patients should be regularly screened for these complications and treatment adherence.
Diabetic ketoacidosis is the most important acute complication of T1D. It may be a result of undiagnosed or undreated T1D, or if a T1D patient does not increase their insulin sufficiently during sickness or stress. Typical features include Kussmaul breathing, hypovolaemia, acetone-breath, reduced level of consciousness, and abdominal pain. Labs show blood glucose > 11 mM, ketosis, and metabolic acidosis. Treatment is by i.v. hydration and rapid-acting insulin. Cerebral oedema may occur if blood glucose correction occurs too rapidly.
Diabetes mellitus type 2
Diabetes mellitus type 2 (T2D) is less common than T1D but becoming more and more prevalent due to obesity epidemic. Unlike T1D patients, T2D patients are often obese and have signs of insulin resistance like acanthosis nigricans.
Treatment is by lifestyle changes and oral antidiabetics like metformin.
17. Obesity and metabolic syndrome
Introduction
Overweightness can be defined as BMI in the 85th – 95th percentile for age and sex and obesity as BMI > 95th percentile for age and sex. More and more children are becoming overweight as part of the obesity epidemic in developed countries. There are many complications of obesity.
Unlike in adults, there is no consensus as to how metabolic syndrome should be defined in children. However, we may define it as in adults, with the presence of three out of these four features:
- Obesity
- Hypertension
- Dyslipidaemia
- Impaired glucose metabolism (diabetes or prediabetes)
We can distinguish two types of obesity according to the cause, simple (exogenous) obesity and secondary (endogenous) obesity.
Etiology
Simple obesity is due to lifestyle factors like increased consumption of energy-dense foods and decreased energy expenditure, as well as genetic factors. This accounts for 95% of cases of childhood obesity.
Secondary obesity is due to medical causes like endocrine disorders, chromosomal disorders, genetic disorders, etc. This accounts for only 5% of obesity cases and can be due to:
- Neuroendocrine
- Hypothyroidism
- PCOS
- Growth hormone deficiency
- Cushing disease
- Immobility
- Muscular dystrophy
- Spina bifida
- Cerebral paresis
- Psychiatric disease
- Depression
- Eating disorder
- Iatrogenic
- Glucocorticoid treatment
- Chromosomal abnormalities
- Down syndrome
- Klinefelter syndrome
- Genetic syndromes
- Prader-Willi syndrome
- Bardet-Biedl syndrome
- Monogenic obesity
- (Only approx. 200 patients are known worldwide)
- Leptin deficiency
- Leptin receptor mutation
- Melanocortin receptor mutation
- POMC mutation
Clinical features
- Skin lesions
- Acanthosis nigricans – hyperpigmentation on neck, axilla. Sign of insulin resistance
- Striae
- Mycosis
- Intertrigo
- Acne
- Hirsutism
Evaluation of obesity
- Later onset, normal or increased linear growth, normal psychomotor development, non-extreme obesity
- -> exogenous obesity most likely
- Beginning early in life (< 6 years), delayed psychomotor development, extreme obesity
- -> suspect genetic disease
- Decreased linear growth (height), decreased bone age
- -> suspect endocrine disease
- Beginning early in life (< 6 years), normal psychomotor development, extreme obesity
- -> suspect monogenic disease
Management of obesity
- Healthier eating (regular meals, eating together with the family, less energy-dense foods)
- More physical activity (walking/biking as transport, playing outdoor, sports)
- Decreased television and small screen use
Drug treatment or bariatric surgery may be used in very severe cases.
Complications of childhood obesity
-
- Cardiovascular
- Dyslipidaemia
- Hypertension
- Atherosclerosis
- Endocrine
- Type 2 diabetes
- Pulmonary
- Sleep apnoea
- Asthma
- Gastrointestinal
- Gallbladder disease
- Non-alcoholic fatty liver disease
- Musculoskeletal
- Slipped capital femoral epiphysis
- Psychosocial consequences
- Poor self-esteem
- Anxiety
- Depression
- Cardiovascular
(Prader-Willi syndrome)
- Due to deletion of paternal genes at 15q11-q13
- Clinical features
- Hypotonia during infancy
- Hypogonadism
- Short stature
- Morbid obesity (due to hyperphagia)
- Poor feeding in the first year of life
- Developmental delay
- Diagnostics
- According to certain diagnostic criteria, based on typical clinical features and genetic testing
18. Microcytic anaemias
Introduction to anaemia in childhood
Anaemia is defined as Hb < 2 standard deviations below the mean for the same age. For each age group, this is approximately:
- Neonate – 140 g/L
- Children 1 months to 1 year – 100 g/L
- Children 1 to 12 years – 110 g/L
- Children 12 to 15 years – 120 g/L
Symptoms are similar as in adults. In case of non-acute anaemia (most types), most children are asymptomatic until the Hb reaches 70 g/L. Typical symptoms include fatigue, pallor (especially of conjunctivae), and poor mental performance.
Iron deficiency anaemia
Iron deficiency anaemia is the most common anaemia in the world and the most common nutritional deficiency in children. It affects up to 10% of adolescent females but <1% of adolescent males.
It may be due to inadequate intake or increased blood loss. Inadequate intake can be from too early introduction of cow’s milk in the infant’s diet, or the use of infant formula which is not iron-fortified. Cow’s milk has low bioavailability of iron and causes occult intestinal blood loss, and should therefore not be introduced into the diet during the first year. Increased blood loss is a common mechanism of iron deficiency in adolescent girls due to menstruation.
Symptoms are as for other anaemias, with the possible addition of pica.
Diagnosis is based on the presence of microcytic hypochromic anaemia and features of iron deficiency, which is most commonly determined by low levels of ferritin (< 15 µg/L).
An empiric trial of oral iron can be tried even in the absence of a definite diagnosis. Oral iron causes a rapid improvement in symptoms, and Hb levels increase within a few weeks. A follow-up appointment is made after a few weeks to determine whether the treatment is effective; if not, other causes of microcytic anaemia should be sought.
Treatment includes dietary advice and oral iron supplement. Most infants receive enough iron from breast milk for the first 6 months, but after month 6 the diet should be supplemented with iron-rich foods or supplements. Menstruating adolescent women should have iron-rich diet or iron supplements. Supplements should be continued for 3 months after Hb has normalised, to refill iron stores.
Thalassaemia
Thalassaemia is characterised by decreased or absent synthesis of one or more globin chains, which leads to abnormal haemoglobin molecules. It’s more common in Mediterranean and Asian populations.
There are two major types of thalassaemia, alpha and beta thalassaemia. In alpha thalassaemia, one or more of the four genes for the alpha globin chain are deleted. In beta thalassaemia, one or both of the genes for the beta globin chain are defective. Severity of the anaemia increases with increased number of affected genes; if all four alpha genes are deleted, the condition is incompatible with life (Hb barts -> hydrops foetalis).
Thalassaemia is can be diagnosed on prenatal DNA analysis. Blood test shows microcytic hypochromic anaemia, evidence of haemolysis, and target cells or teardrop cells on blood smear. The diagnosis is confirmed by haemoglobin electrophoresis or genetic studies.
In mild thalassaemias, no treatment is necessary. In severe ones, lifelong regular blood transfusions are necessary. Treatment with iron chelators are necessary to prevent iron overload. Haematopoietic stem cell transplantation could be curative in severe cases.
Other causes of microcytic anaemia
- Anaemia of chronic disease
- Lead poisoning
19. Normocytic anaemias (haemolytic anaemias)
Sickle cell disease
Sickle cell disease is the most common intrinsic haemolytic anaemia, i.e. the most common cause of haemolytic anaemia due to intrinsic defects of the RBCs. It’s most common in African and Mediterranean populations.
It’s caused by a point mutation in the beta globin gene causes haemoglobin to precipitate into a sickle-shape when deoxygenized. This leads to microvascular occlusion and haemolysis.
Sickle cell disease causes moderately severe anaemia with clinically detectable jaundice due to haemolysis. It also causes hyposplenism, which leads to increased susceptibility to severe infections by encapsulated bacteria, like osteomyelitis and sepsis.
Acute complications can also occur. Acute splenic sequestration crises can occur, where a large number of sickled clls accumulate in the spleen, causing sudden drop in blood volume and splenomegaly. Acute aplastic crisis occurs in case of parvovirus B19 infection, in which the virus causes complete but temporary cessation of RBC production. Vaso-occlusive crises is a consequence of sudden vaso-occlusion, leading to episodes of severe bone pain and dactylitis, as well as priapism.
Disckle cell disease causes a normocytic haemolytic anaemia. Blood smear may show sickle cells. Haemoglobin electrophoresis confirms the diagnosis.
All affected children should be immunised against pneumococci, meningococci, and HiB. Hydroxyurea/hydroxycarbamide can prevent acute crises, but may cause bone marrow suppression. Painful crises should be treated with sufficient analgesics, sometimes requiring opioids. Bone marrow transplant may be used in very severe cases.
G6PD deficiency
Glucose 6-phosphate dehydrogenase (G6PD) deficiency is a deficiency of the rate-limiting enzyme of the pentopse phosphate pathway, which is essential for preventing oxidative damage to RBCs. Deficient RBCs are suspectible to haemolysis when exposed to oxidants. It’s an X-linked condition.
G6PD deficiency can cause neonatal jaundice. Other than this, most patients are asymptomatic until they’re exposed to factor which precipitates oxidative injury and resulting haemolysis. This can occur due to infection, fava beans, and certain drugs like sulphonamides. During these haemolytic crises, Hb drops rapidly, possible falling below 50 g/L over 1 – 2 days. Symptoms of crisis include jaundice, pallor, dark urine, and abdominal or back pain.
G6PD deficiency causes normocytic haemolytic anaemia. On blood smear, bite cells and Heinz bodies may be present. Measurement of the G6PD enzyme activity in RBCs confirms the diagnosis.
Management involves avoiding triggers of haemolytic crises.
Hereditary spherocytosis
Hereditary spherocytosis is the most common cause of intrinsic haemolytic anaemia in Caucasians. It’s caused by mutations in RBC membrane proteins which cause the cells to become spherical in shape and prone to destruction in the spleen.
The severity of anaemia is highly variable, and can range from asymptomatic to severe. Aplastic crisis can occur in case of parvovirus B19 infection.
Diagnosis is based on the presence of spherocytes on the blood smear or based on specific tests. There’s no treatment, but patients should receive folic acid supplements as they have a higher requirement. In case of very severe symptoms, splenectomy may be performed after the age of 7, but only after the child has been immunised against encapsulated bacteria.
20. Macrocytic anaemias (congenital aplastic and hypoplastic anaemias, megaloblastic anaemia)
Fanconi anaemia
Fanconi anaemia is the most common congenital aplastic anaemia, but it is still rare overall (1:100 000). It’s an autosomal recessive disorder caused by mutations in genes responsible for DNA repair.
Most affected children have congenital abnormalities like short stature, abnormal arms and fingers, and microphthalmia. However, as an aplastic anaemia, the characteristic feature is pancytopaenia, which may not become apparent until the age of 5. There’s an increased risk of acute leukaemia.
The recommended treatment is bone marrow transplantation.
Diamond-Blackfan anaemia
Diamond-Blackfan anaemia, also called congenital hypoplastic anaemia, is a rare congenital red cell aplasia which presents during childhood. It causes macrocytic anaemia and is associated with congenital abnormalities of the face, neck, thumbs, etc. The treatment is glucocorticoids and blood transfusions. Bone marrow transplant may also be an option.
Megaloblastic anaemia
Megaloblastic anaemia is a subtype of macrocytic anaemia characterised by decreased DNA synthesis in RBC precursors, causing them to be larger and have characteristic “megaloblastic” features. It’s caused by dietary folate and B12 deficiency, which are relatively rare in children.
It can occur as a result of Crohn disease, pernicious anaemia, malnutrition, etc. Labs show macrocytic anaemia with hypersegmented neutrophils. Serum homocysteine and methylmalonic acid (MMA) are elevated.
Treatment is be treating the underlying cause, and oral supplements.
21. Leukocyte diseases (neutropaenias, neutrophils, eosinophils, monocytosis, lymphocytosis)
Neutropaenia
Neutropaenia refers to a neutrophil count < 1500/µL. The lower the count, the higher the risk for infection. At < 1000/µL there is a significant increase in risk. Most patients do well with > 500/µL, and serious infections occur < 200/µL.
With significant neutropaenia comes recurrent infections by bacteria and certain fungi, mostly endogenous bacteria like s. aureus, e. coli, candida spp. Oral cavity ulcers may also develop.
Some conditions cause congenital neutropaenia, but these are rare. These include:
- Neonatal alloimmune neutropaenia – Transient neutropaenia of newborn due to maternal anti-neutrophil antibodies
- Severe congenital neutropaenia (Kostmann syndrome) – Genetic mutation causes promyelocytes to fail to mature
- Cyclic neutropaenia
- Shwachman-Diamond syndrome – characterised by pancreatic insufficiency + panmyeloid dysfunction
Acquired neutropaenias are more common. These can be due to:
- Sepsis
- Acute leukaemia
- Drugs (antithyroids, cytotoxics)
- Aplastic anaemia
- Malnutrition
The aim should be to treat the underlying condition. In some cases G-CSF and bone marrow transplant may be used.
Neutrophilia
Neutrophilia refers to neutrophils > 8000/µL. Neutrophilia is usually a sign of infection, especially bacterial, but it can also be due to other causes:
- Inflammatory diseases
- IBD
- Juvenile RA
- Leukaemia
- Myeloproliferative neoplasms
- Congenital disorders
- Leukocyte adhesion deficiency
Eosinophilia
Eosinophilia refers to eosinophils > 810/µL. This is typically a sign of helminth infection or allergy:
- Allergy
- Atopic dermatitis
- Asthma
- Allergic rhinitis
- Food allergy
- Helminth infection
- Leukaemia
- Hodgkin lymphoma
Monocytosis
Monocytosis refers to monocytes > 1000/µL. It’s not common, and I can’t find any common reasons for monocytosis.
Lymphocytosis
Lymphocytosis refers to lymphocytes > 4500/µL. It’s usually a sign of viral infections, but also toxoplasmosis and pertussis.
- Infectious mononucleosis
- Toxoplasmosis
- Viral infections
- Pertussis
22. Immune thrombocytopaenias
Disorders of platelets are most commonly associated with bleeding in the skin and mucosa, while bleeding disorders most commonly cause bleeding into joints or muscle.
Immune thrombocytopaenia
Immune thrombocytopaenia (ITP), also called immune thrombocytopaenic purpura or idiopathic thrombocytopaenia, is an immune-mediated cause of thrombocytopaenia in children. It’s the most common cause of thrombocytopaenia in childhood.
It’s caused by the formation of IgG antibodies against platelets. This typically occurs a few weeks after a viral infection or vaccination, but it can also be secondary to malignancy or autoimmune disease.
Symptoms develop over days and include petechiae, purpura, bruising, and epistaxis. Intracranial bleeding is a serious but rare complication.
It’s a diagnosis of exclusion, so we must exclude other causes of thrombocytopaenia like leukaemia, aplastic anaemia, and congenital disorders. In ITP, there is isolated thrombocytopaenia, and the other cell lines are normal. The platelet count can be very low, even < 10 G/L (normally 140 – 440).
Most cases are unproblematic and spontaneously resolve after a few weeks. If there is severe bleeding, glucocorticoids or IVIG as well as platelet transfusion can be used. Children should avoid trauma and contact sports until it resolves. 10-20% develop chronic ITP.
Neonatal alloimmune thrombocytopaenia
Neonatal alloimmune thrombocytopaenia occurs when the mother produces antibodies against the foetuses platelet antigens. It can be anywhere from asymptomatic or severe, potentially causing intracranial haemorrhage. Treatment is glucocorticoids or IVIG as well as platelet transfusion, if necessary.
23. Haemorrhagic diseases (von Willebrand disease, haemophilias)
von Willebrand disease
von Willebrand disease (vWD) is caused by a deficiency of von Willebrand Factor (vWF), which is involved in adhesion of platelets to each other and to damaged blood vessels. It also prevents breakdown of factor VIII, a clotting factor. vWD is the most common bleeding disorder and is autosomal dominant.
The severity of the symptoms depend on the degree of deficiency of the factor. The most common symptoms are bruising, excessive bleeding after surgery, epistaxis, and menorrhagia.
Labs can measure the vWF activity to make the diagnosis. In mild cases, desmopressin treatment is usually enough. In more severe cases, plasma-derived factor VIII concentrate must be given (as it contains vWF as well).
Haemophilia
Haemophilia is the most common severe bleeding disorder. It’s an X-linked disorder, and there are two types, haemophilia A (factor VIII deficiency) and haemophilia B (factor IX deficiency). Haemophilia A is most common.
Like for vWD, the severity of the symptoms depend on the degree of deficiency of the factor. In mild or moderate cases, there’s an increased tendency to bleed after mild trauma or surgery. Trauma may cause haemarhthosis or soft tissue haematomas. In severe cases, there may be recurrent spontaneous bleeding into joints and muscles, and bruising from trivial pressure, like being picked up. Recurrent haemarthrosis destroys the joint.
Labs can measure the FVIII or FVIX activity to make the diagnosis. Treatment is by regular, prophylactic recombinant clotting factor infusions, which reduces the bleeding tendency and risk for joint damage. Parents and eventually the child itself can be taught to administer the infusion, which is given 1 – 3 times per week. Extra infusions should be given in case of acute bleeding episodes. In some cases, the patient begins to produce antibodies against the recombinant factors, which decrease or completely inhibit the effects of treatment.
Desmopressin stimulates release of FVIII, and may be used alone to manage mild haemophilia A without the use of clotting factor infusions.
24. Leukaemias (ALL, AML, CML)
Acute lymphoblastic leukaemia
Acute lymphoblastic leukaemia (ALL) is the most common childhood cancer, and it accounts for 80% of childhood leukaemia. The presentation peaks at 2 – 5 years of age. ALL may be originate from B-cells (most common) or T-cells. ALL is much more common in people with Down syndrome.
Acute leukaemia may present with bone marrow failure (pallor, recurrent infections, bleedings), hepatosplenomegaly, and/or lymphadenopathy, often with non-specific signs like fever, malaise, fatigue, and weight loss. Relapsed acute leukaemia may present with infiltration of the CNS or testes.
Lab tests show anaemia, thrombocytopaenia, and either low, normal, or high leukocyte count. Blood smear or flow cytometry can show blast cells in the peripheral blood. For definitive diagnosis, bone marrow examination must be made. After diagnosis, lumbar puncture and CSF analysis must be made to look for CNS involvement.
The management of ALL is chemotherapy, which cures 90% of cases. Induction therapy is the first stage, which eradicates the leukaemic blasts. Then, moderate intensity chemotherapy is continued for up to 3 years. Intrathecal chemotherapy prevents CNS relapse. During chemotherapy, patients should receive TMP-SMX prophylaxis for PCP.
Acute myeloid leukaemia
Acute myeloid leukaemia (AML) accounts for 20% of childhood leukaemias. It’s survival is lower than that of ALL. Presenting symptoms and evaluation are similar as for ALL. Treatment is also chemothereapy, but with different drugs.
Chronic myelogenous leukaemia
Chronic myelogenous leukaemia (CML) is rare in childhood, accounting for < 5%. Symptoms include B symptoms and splenomegaly.
25. Lymphomas (Hodgkin, non-Hodgkin)
Hodgkin lymphoma
Hodgkin lymphoma (HL) is more frequently seen in older children. About 80% can be cured, and the cure rate is high even for those with disseminated disease.
It presents with painless lymphadenopathy, most frequently in the neck. The lymph nodes may cause obstruction of the airway or superior vena cava. B symptoms may also occur. The clinical history is often long (several months).
Lymph node biopsy, radiological assessment of all lymph node sites, and bone marrow biopsy are necessary for the diagnosis and to determine the stage. PET scan is often useful.
Treatment is by chemotherapy with or without radiotherapy.
Non-Hodgkin lymphoma
Non-Hodgkin lymphoma (NHL) is more frequently seen in younger children. About 80% can be cured here as well.
NHL may present with a mediastinal mass which obstruct airways or the SVC, or with painless lymphadenopathy in the head, neck, or abdomen. Enlarged lymph nodes in the abdomen may cause intenstial obstruction or intussusception.
Lymph node biopsy, radiological assessment of all lymph node sites, and bone marrow biopsy are necessary for the diagnosis and to determine the stage. PET scan is often useful.
Treatment is by chemotherapy.
26. Brain tumours in childhood
Brain tumours are the most common solid tumours in children, and the second most common malignancy overall. Unlike in adulthood, CNS tumours in childhood are almost always primary.
The most common types are:
- Astrocytoma – varies from low grade to high grade (glioblastoma)
- Medulloblastoma – in the midline of the posterior fossa
- Ependymoma – mostly in 4th ventricle
- Brainstem glioma – very malignant, poor prognosis
- Craniopharyngoma – a remnant of the Rathke pouch
- Atypical teratoid/rhabdoid tumour – rare, most commonly in young children
CNS tumours can present with many different symptoms:
- Symptoms of mass effect -> increased ICP
- Vomiting (especially right after waking up in the morning)
- Headache
- Seizures
- Symptoms of cranial nerve impingement
- Visual defects
- Abducent palsy
- Symptoms of CSF obstruction -> hydrocephalus
- In young children with open sutures -> enlarged head, bulging fontanelles
- Symptoms of pituitary involvement
- Precocious puberty
- Diabetes insipidus
- Symptoms of cerebellar involvement
- Ataxia
- Nystagmus
MRI is the best modality to evaluate CNS tumours. Lumbar puncture may be performed to look for CNS involvement, but not if there’s increased ICP.
The main treatment is surgery, which aims to resect as much as possible, to provide specimens for pathohistology, and to alleviate symptoms. Adjuvant chemo and/or radiotherapy is often used. Dexamethasone can be used to decreased ICP.
27. Neuroblastoma and Wilms tumor
Neuroblastoma
Neurblastoma is a tumour of neural crest cells in the adrenal medulla and sympathetic nervous system. It may be anywhere from benign to very malignant, and mostly affects children < 5.
The primary tumour may be anywhere along the sympathetic chain, but most commonly in the abdomen. It may present as an incidentally discovered mass which or due to symptoms like abdominal pain or constipation. The mass is irregular and crosses the midline, in contrast to Wilms tumour. Symptoms of metastases include periorbital ecchymoses, exophthalmos, bone pain, and anaemia. Opsomyoclonus is a characteristic but rare paraneoplastic syndrome characterised by myoclonus and rapid involuntary eye movements.
The diagnosis is likely when imaging shows calcified tumour and there are catecholamine metabolites in the urine, but definitive diagnosis requires biopsy. MIBG and bone marrow sampling are performed to check for metastasis.
In many infants, neuroblastoma resolves spontaneously. However, in older children, the prognosis is not so good despite treatment. A combination of surgery, radio, and chemo are often used.
Wilms tumour
Wilms tumour, also called nephroblastoma, is a tumour of the metanephric blastema. It’s the most common renal tumour in childhood. It mostly presents before the age of 5. 5% of cases are bilateral.
It commonly presents as a painless, incidentally discovered abdominal mass, sometimes with haematuria. Unlike in neuroblastoma, this abdominal mass does not cross the midline.
Ultrasound or CT/MRI shows a characteristic renal mass. As part of the preoperative evaluation, it’s important to assess for lung metastases and the function of the contralateral kidney.
Treatment is usually by neoadjuvant chemotherapy followed by surgery (nephrectomy).
Wilms tumour may be part of a syndrome called WAGR syndrome, characterised by Wilms tumor, aniridia, genitourinary anomalies, and retardation (intellectual disability).
28. The types of dehydration and their treatment
(Maintenance fluid requirement)
For first week of life:
- Children 3,5 – 10 kg
- 100 mL/kg/day
- 4 mL/kg/hour
- Children 10 – 20 kg
- 1000 mL + 50 mL/kg/day for every kg over 10 kg
- 2 mL/kg/hour
- Children > 20 kg
- 1500 mL + 20 mL/kg/day for every kg over 20 kg
- 1 mL/kg/hour
Dehydration
Infants are at higher risk for dehydration than older children and adults because:
- A larger percentage of their body weight is water (75% in term infants, higher in preterms)
- They have higher metabolic rate
- They have increased surface area-to-body mass ratio, which allows greater water loss through the skin
- Their kidneys may not be fully mature -> decreased ability to concentrate urine -> water loss in urine
The most common causes of dehydration are:
- Gastroenteritis
- Burns
- Sepsis
- Diabetic ketoacidosis
- Diabetes insipidus
- Nephrotic syndrome
- Painful swallowing (due to sore throat, fever, etc.)
The assessment of the severity of the dehydration is based on clinical evaluation, and is important in determining the treatment:
| Signs | Mild | Moderate | Severe (shock) |
| Fluid loss (% of body weight lost as fluid) | < 5% | 5 – 10% | > 10% |
| General status and behaviour | Thirsty, alert | Thirsty, restless, irritable | Drowsy, limp, shock |
| Pulse | Normal | Weak | Decreased |
| Tachycardia | Absent | Present | Present |
| Blood pressure | Normal | Normal or orthostatic hypotension | Decreased |
| Turgor | Normal | Slightly decreased | Markedly decreased |
| Eyes | Normal | Sunken | Markedly sunken |
| Anterior fontanelle | Normal | Sunken | Markedly sunken |
| Buccal mucosa | Normal or slightly dry | Dry | Very dry, parched |
| Capillary refill | Normal | Normal | Prolonged (> 3 s) |
| Urine output | Normal | Oliguria | Anuria or severe oliguria |
We can classify the types of dehydration according to the plasma tonicity:
- Hypotonic (Na+ < 130 mM)
- Isotonic (Na+ 130 – 150 mM)
- Hypertonic (Na+ > 150 mM)
Fluid therapy
In case of mild-moderate dehydration, oral rehydration therapy (ORT) with oral rehydration salts (ORS, e.g. WHO ORS) should be used rather than i.v. fluids, as they’re cheaper, safer, and easier to administer. However, this is not an option in case of persistent vomiting, impaired mental status with risk of aspiration, or ileus.
In case of moderate-severe dehydration, fluid resuscitation with intravenous fluids are necessary. A rapid infusion or bolus of 20 mL/kg crystalloid (saline or Ringer) is given and the response is assessed. If there is response, the bolus should be repeated until adequate perfusion is restored. If there is not response, the child may require intensive therapy and mechanical ventilation. After adequate perfusion has been restored (shock corrected), correction of the remaining fluid deficit can usually be achieved with oral rehydration therapy rather than continued i.v. therapy.
When managing dehydration in children, it’s important to replace not only the lost fluid but the daily maintainance fluid requirement as well. For example, for a 30 kg child with severe (10%) fluid loss:
- Maintenance fluid requirement = 1500 mL + (20 mL * 10 kg) = 1700 mL
- Fluid deficit = 10% of 30 kg = 10% of 30 L = 3000 mL
- Total fluid requirement = 1700 + 3000 = 4700 mL
After the patient is no longer in shock (or if the patient was never in shock), the total calculated fluid requirement should be given over 24 hours. Of the fluid deficit, the first half is given in the first 8 hours, and the other half is given in the next 16 hours. The maintenance fluid requiriment, should be distributed evenly in the first 24 hours.
If there is a concomitant sodium imbalance, the dehydration should be corrected first, followed by correction of the sodium imbalance.
29. Hyponatremia/hypernatremia and hypokalaemia/hyperkalaemia
Hyponatraemia
Hyponatraemia is defined as:
- Mild: Na+ 130 – 135 mM
- Moderate: Na+ 120 – 130 mM
- Severe: Na+ < 120 mM
- Acute hyponatraemia: developing within 48 hours
- Chronic hyponatraemia: developing over > 48 hours
We classify hyponatraemia according to the volume status it accompanies. It can be hypovolaemic, euvolaemic, or hypervolaemic.
- Hypovolaemic hyponatraemia – is most commonly a consequence of a dehydrated child being managed with water.
- Euvolaemic hyponatraemia – is usually a result of SIADH or water intoxication. SIADH can occur due to lung disease (pneumonia, mechanical ventilation) or CNS disease (injury, infection, tumour).
- Hypervolaemic hyponatraemia – can be due to kidney failure, liver failure, or heart failure.
The presence and severity of symptoms depend on the severity of the hyponatraemia, as well as how quickly it develops. The brain can adapt to hyponatraemia, but these adaptive mechanisms take at least a day to kick in. For this reason, acute hyponatraemia is usually symptomatic.
Most symptoms are due to cerebral oedema, and initially include nausea and malaise, but may develop into lethargy, seizures, and coma if the sodium level continues to fall.
Management is by correcting the hyponatraemia, but not too quickly, as this can cause CNS demyelination, which is irreversible. In many cases, treating the underlying cause restores normal siodium status without the need for saline infusion. If SIADH is the cause, fluid restriction helps. However, if hyponatraemia is severe or symptomatic, it should be corrected. Intravenous isotonic saline may be used in less severe cases, with i.v. hypertonic saline being reserved for more severe cases.
Hypernatraemia
Hypernatraemia is defined as Na+ > 150 mM, and is less common than hyponatraemia in children. It’s often accompanied by dehydration.
Hypernatraemia occurs if there is water or hypotonic fluid loss which is not replaced or if there’s excessive salt water intake. In children, this is most commonly due to:
- Gastroenteritis (especially rotavirus) without fluid replacement
- Diabetes insipidus
- Inadequate water intake
- Iatrogenic excess sodium administration
Symptoms vary from irritability, restlessness, vomiting, to severe neurological symptoms like lethargy, seizures, and coma, depending on the acuity and severity.
Treatment involves i.v. administration of isotonic fluids and treatment of the underlying cause. The correction rate should not make serum Na+ decrease more than 12 mM/L every 24 hours to prevent brain oedema.
Hypokalaemia
Hypokalaemia is defined as K+ < 3,5 mM.
Increased gastrointestinal loss is the most common cause of hypokalaemia in children, including diarrhoea and vomiting. Other causes include alkalosis, and drugs like insulin, salbutamol, and diuretics.
Symptoms vary from muscle weakness and paralysis, to cardiac arrhythmias, depending on the acuity and severity. It’s also associated with ECG changes like U waves, T wave flattening, and ST depression.
Treatment is by correction. Severe or symptomatic hypokalaemia should be corrected urgently with i.v. potassium chloride. In less severe cases, oral potassium therapy is usually sufficient.
Hyperkalaemia
Hyperkalaemia is defined as K+ > 5,5 mM.
The causes are mostly the same as in adults:
- Renal disease (AKI or CKD)
- Rhabdomyolysis
- Tumour lysis syndrome
- Acidosis
- RAAS inhibitor drugs
- Pseudohyperkalaemia (haemolysis of the blood sample)
Most children have mild or moderate hyperkalaemia, and are therefore asymptomatic. In severe hyperkalaemia, muscle weakness and cardiac arrhythmias can develop. It’s also associated with ECG changes like peaked T waves and wide QRS.
In severe or symptomatic hyperkalaemia, it should be corrected emergently. Emergent treatment of hyperkalaemia involves causing intracellular shift of potassium. This can be achieved by inhaled salbutamol, i.v. calcium, or i.v. insulin + glucose.
Following resolution of the symptoms or severe hyperkalaemia, we must remove excess potassium from the body. This can be achieved by K+-losing diuretics (loop or thiazides), K+-binding resins, or dialysis.
30. Vomiting in infancy and childhood
Vomiting refers to the forceful expulsion of gastric contents which is preceded by nausea and accompanied by gagging and retching. It’s a common problem in children, and there are many possible causes.
Gastroesophageal reflux
It’s important to differentiate vomiting from physiologic gastroesophageal reflux (GER), also called “spitting up”. This occurs in virtually all infants, but usually stops after 1 year. In contrast to vomiting, the stomach content in GER is not forcefully ejected, but it rather spills out of the mouth effortlessly.
Differential diagnosis of vomiting in neonates and infants
| Cause | Distinguishing features |
|---|---|
| Pyloric stenosis | First month of life, projectile vomiting immediately after feeding, visibly distended stomach, visible peristalsis |
| Intestinal atraesia | Vomit may be bilious, abdominal distension |
| Metabolic disorders | Hepatomegaly, abnormal glucose/electrolyte metabolism, jaundice |
| Gastroenteritis (usually viral) | Acute onset, also has diarrhoea |
| Milk or soy allergy | Formula-fed infant, allergic symptoms |
| Increased ICP | Bulging fontanelle, papilloedema, neurologic symptoms |
| Severe infection (sepsis) | Hypothermia/fever, respiratory distress, tachycardia, neurologic symptoms |
| GERD | Irritability, feeding problems (nonspecific symptoms only) |
| Adrenal insufficiency (CAH) | Projectile vomiting, hyponatraemia, hyperkalaemia, metabolic acidosis, hypotension |
| Hirschprung disease | Abdominal distension, failure to pass stool/meconium |
Differential diagnosis of vomiting in older children
| Cause | Distinguishing features |
|---|---|
| Gastroenteritis (usually viral) | Acute onset, also has diarrhoea |
| GERD | Irritability, feeding problems (nonspecific symptoms only) |
| Pharyngitis | Sore throat, fever, dysphagia, exudate on tonsils |
| Food poisoning | During travel or after eating out, others who ate the same have symptoms |
| Increased ICP | Papilloedema, neurologic symptoms, morning vomiting |
| Cyclic vomiting syndrome | Recurrent attacks of severe vomiting, no anatomical anomalies |
| Appendicitis | Acute abdomen, pain at McBurney point |
| Mechanical bowel obstruction | Acute abdomen, bilious vomiting |
| Otitis media | Ear pain, bulging ear drum, fever |
| Pregnancy | Older adolescent female, nausea in morning, amenorrhoea |
Red flags of vomiting
- Bilious vomit -> Intestinal obstruction
- Abdominal pain -> peptic ulcer, appendicitis
- Haematemesis -> peptic ulcer, Mallory-Weiss tear
- Early morning vomiting -> increased ICP (or pregnancy)
- Haematochezia -> intussusception
- Headache -> migraine
- Marked abdominal distension -> intestinal obstruction
Examination of child with vomiting
Evaluating the hydration status is important, to rule out dehydration. The abdomen should be examined for masses, distension, organomegaly, tenderness, and guarding.
Lab evaluation should include electrolytes, CBC, pancreatic enzymes, kidney function tests, and liver function tests. Ultrasound and CT may be very helpful in determining the etiology.
31. Acid-base balance
Backround physiology review
Normal values:
- Blood pH = 7,35 – 7,45
- HCO3– = 22 – 28 mEq/L
- pCO2 = 35 – 45 mmHg
- Base excess = -5 – +5
pH is generally only surviveable in the 6,8 – 8,0 range.
If the compensatory mechanisms return pH into normal range, there is complete compensation. If still outside normal range, there is partial compensation. Compensatory mechanisms for pH changes:
- Bicarbonate buffer
- Phosphate buffer
- Protein buffer
- Respiratory compensation (CO2 excretion)
- Renal compensation (H+ excretion)
Definitions:
- Acidosis = process which decreases pH
- Alkalosis = process which increases pH
- Acidaemia = pH < 7,35
- Alkalaemia = pH > 7,45
- Anion gap = [Na+] – ([Cl–] + [HCO3–])
- Normally 10 – 14 mEq/L
Metabolic acidosis
Metabolic acidosis is defined as acidosis in the presence of HCO3 < 22 mEq/L. It’s a common occurence in very ill and hospitalised children.
We distinguish normal anion gap metabolic acidosis (NAGMA), which is generally caused by increased loss of bicarbonate or decreased loss of H+, from high anion gap metabolic acidosis (HAGMA), which is generally caused by elevated levels of acids, like lactic acid, ketone bodies, etc. The most common causes of each are:
- Normal anion gap metabolic acidosis
- Diarrhoea
- Renal tubular acidosis
- High anion gap metabolic acidosis
- Lactic acidosis (shock)
- Ketoacidosis (diabetes, alcoholism)
- Kidney failure
- Poisoning
- Ethylene glycol
- Methanol
- Salicylates
- Inborn errors of metabolism
Metabolic acidosis itself is usually asymptomatic as long as the acidaemia isn’t severe. The underlying cause is what usually produces most of the symptoms. Severe acidaemia causes impaired cardiac contractility, increased risk of arrhythmias, and vasoconstriction in the pulmonary circulation, which may worsen the underlying condition. The characteristic Kussmaul breathing occurs only in moderate or severe acidosis. Chronic metabolic acidosis causes failure to thrive.
Treatment is mostly targeted at the underlying disease. Intravenous bicarbonate therapy may be used in severe cases.
Metabolic alkalosis
Metabolic alkalosis is defined as alkalosis in the presence of HCO3– > 26 mEq/L.
We can distinguish chloride responsive metabolic alkalosis, which reponds to fluids containing chloride (sodium chloride or potassium chloride), and chloride resistant metabolic alkalosis, which doesn’t. The most common causes are:
- Chloride responsive metabolic alkalosis
- Vomiting (most common cause overall)
- Nasogastric suction
- Diuretics
- Cystic fibrosis
- Chloride resistant metabolic alkalosis
- Cushing syndrome
- Hyperaldosteronism
- Rare syndromes (Bartter, Gitelman, Liddle)
Metabolic alkalosis itself is usually asymptomatic as long as the alkalaemia isn’t severe. The underlying cause or the associated electrolyte disturbances are what usually produces most of the symptoms. Hypokalaemia is often present and can cause symptoms. Severe alkalaemia can cause arrhythmias, hypoventilation, and decreased cardiac output.
Chloride responsive metabolic alkalosis responds to volume repletion with sodium and potassium chloride, which also corrects the hypokalaemia. As always, treatment of the underlying cause is important. Administration of acid is rarely necessary.
Respiratory acidosis
Respiratory acidosis is defined as acidosis with pCO2 > 45 mmHg (hypercapnia). The most common causes are:
- CNS depression (encephalitis, opioids)
- Disorders causing paralysis of respiratory muscles (botulism, Guillain-Barré syndrome, muscular dystrophy)
- Lung disease (pneumonia, asthma)
- Laryngospasm
Treatment is targeted at the underlying disease. Mechanical ventilation may be necessary.
Respiratory alkalosis
Respiratory alkalosis is defined as alkalosis with pCO2 < 35 mmHg. The most common causes are:
- Hypoxaemia (CO poisoning, cyanotic heart disease)
- Central stimulation (anxiety or brain tumour)
- Mechanical ventilation
- Hyperammonaemia
Treatment is targeted at the underlying disease. Breathing into a bag might help in case of anxiety.
32. Antibiotic therapy in the infancy and childhood
Choice of antibiotic
Selecting the correct antibiotic treatment is important. It depends on multiple factors:
- The site of infection
- The clinical features
- The host immunity
- Likely causative agents
- Local epidemiology of resistance
- The pharmacokinetics and pharmacodynamics of the antibiotics
- The pathogen’s susceptibility to antibiotics (if known)
In most cases, antibiotic therapy begins with empirical antibiotics, which are usually broad-spectrum. These are chosen based on the most likely causative bacteria and the local epidemiology. This choice is usually based on guidelines, either local or international.
In case of many infections, empirical antibiotics is usually enough to treat the condition. This is the case for infections like acute otitis media. Because of the high probability of empirical antibiotics being enough in these cases, cultures are rarely performed. However, if empirical antibiotic therapy fails, culture should be made, as an atypical pathogen may be the cause.
In case of other infections, usually more severe ones, empirical therapy is not initiated until culture specimens (blood, urine, sometimes CSF) have been obtained. One exception is sepsis, in which case empiric antibiotic therapy must be started as early as possible.
Bacterial cultures give the causative pathogen as well as its sensitivity to various antibiotics. If culture was made and the patient is still taking empirical antibiotics when the culture result is ready (takes a few days), we switch the patient over to definitive, specific antibiotic therapy, consisting of narrow-spectrum antibiotics which the pathogen is sensitive to. This reduces the side effects of antibiotic therapy.
In some cases, the clinical features are so characteristic, or there are rapid point-of-care tests available, which allow for immediate diagnosis of the causative pathogen, which in turn allows for immediate initiation of definitive, specific antibiotic therapy. This is the case for streptococcal pharyngitis, for example.
Immunocompromised children may have infections by unusual bacteria which may require other antibiotics or more intensive antibiotic therapy than in immunocompetent ones.
It’s important to choose antibiotics against which achieve therapeutic levels at the site of infection. As an example, ceftriaxone has particularly good CNS penetration compared to other 3rd generation cephalosporins. Aminoglycosides are much less effective in sites with low pH and oxygen tension like abscesses.
Dosage and administration of antibiotics in childhood
Most antibiotics used in childhood are dosed according to the patient’s weight. As such, it’s important to know this parameter before prescribing antibiotics.
In general, the least invasive method of administration which is appropriate is chosen. In case of severe infections, antibiotics are usually administered intravenously in the first days, followed by conversion to oral therapy. If topical therapy is possible, it should be preferred over oral.
Contraindicated antibiotics in childhood
- Fluoroquinolones -> cartilage damage
- Tetracyclines -> teeth discoloration
- Chloramphenicol (for infants) -> grey baby syndrome
Most common paediatric conditions in which antibiotics are prescribed
| Condition | Antibiotic of choice |
|---|---|
| Acute otitis media | Amoxicillin or amoxicillin-clavulanic acid |
| Bacterial rhinosinusitis | |
| Neonatal sepsis | Ampicillin + gentamycin or ampicillin + cefotaxime |
| Neonatal bacterial meningitis | |
| Sepsis (non-neonate) | Ceftriaxone |
| Pneumonia (CAP) | Penicillin or amoxicillin |
| Streptococcal pharyngitis | Penicillin or amoxicillin |
| Urinary tract infection | 2nd/3rd gen cephalosporin, or aminoglycoside |
Unfortunately, antibiotics are often prescribed on poor indications, such as diarrhoea or fever without focus. This leads to unnecessary antibiotic use.
33. Fever in childhood (causes, measurement, antipyretics)
Measurement of temperature in childhood
Rectal thermometry is the gold standard for temperature evaluation. It is generally used for infants and children too young to take oral temperature. > 38,0°C is considered fever. However, there are some contraindications to rectal thermometry. Contraindications to rectal thermometry in neonates include:
- Neutropaenia
- Bleeding diathesis (like thrombocytopaenia)
- Necrotising enterocolitis
Axillary, temporal artery, and tympanic measurements are less reliable. There are a few contraindications to rectal thermometry, in which case axillary measurement or other measurement could be used. Non-rectal temperatures are consistently lower than rectal temperatures, and are usually approx 0,5°C lower, but the difference generally varies too much for a standard conversion to rectal temperature. However, if non-rectal temperature is > 38°C the rectal temperature must be either the same or higher.
We can distinguish multiple “severities” of fever based on the temperature:
- Normal body temperature – 37°C (range 36 – 37,5°C)
- Subfebrile – 37,5 – 38°C
- Fever – > 38°C
The majority of febrile children will have no obvious findings of infection during physical examination, but we must still perform it. Because of this, we must rely on a combination of history, physical examination, and diagnostic tests to determine the risk of serious infection.
Differential diagnosis of fever in neonates
Neonatal fever (and fever in any < 3 month old) is always an indication for inpatient evaluation of infection and empiric antibiotics. Note that neonates can also develop hypothermia as a sign of severe infection. Also important to know the most common causative pathogens for neonatal infection, GBS, E.coli, Listeria, and Klebsiella.
| Etiology | Typical findings |
| Neonatal invasive disease (sepsis, meningitis, pneumonia) | Irritability, lethargy, tachycardia, respiratory distress, shock. Seizures (meningitis) |
| Meningococcal disease | Above + purpuric rash |
| Urinary tract infection | Vomiting, jaundice |
| Bacterial gastroenteritis | Diarrhoea |
| Osteomyelitis | Decreased use of limb, local swelling and erythema |
| Septic arthritis | Swollen, painful, warm, red joint |
| Omphalitis | Signs of infection (pus, erythema) around umbilicus |
| Herpes simplex virus (HSV) infection | Mucocutaneous vesicles, seizures, focal neurological symptoms, conjunctivitis |
Differential diagnosis of fever in children
Fever in childhood is usually due to viral infection, so antibiotics are usually not indicated. However, fever for more than 3 days is suspicious for bacterial infection (or Kawasaki) and should be investigated as one.
| Etiology | Typical findings |
|---|---|
| Viral URTI | Nasal congestion, nasal discharge, cough. Child does not appear severely ill |
| Kawasaki disease | Conjunctivitis, red cracked lips, polymorphic lips, erythema and oedema on hands |
| Pneumonia | Cough, respiratory distress, poor feeding, irritability |
| Tonsillitis | Sore throat, vomiting, dysphagia, exudate on tonsils |
| Viral exanthems | Rash |
| Sepsis | Shock, skin bleedings, ill appearance, altered mental status |
| UTI | Irritability, poor feeding, poor weight gain, urinary symptoms |
| Meningitis | Irritability, poor feeding, meningeal signs (if older), purpuric rash (if meningococcal) |
| Endocarditis | Malaise, anaemia, embolic phenomena, new heart murmur |
Antipyretics in paediatrics
The most commonly used antipyretics in childhood are paracetamol and ibuprofen. The dose for each are:
- Paracetamol
- Doses are up to 3x a day
- (15 mg/kg every 4 – 6 hours, max 75 mg/kg/day)
- 250 mg up to 3x a day for children 3 – 7 years
- 500 mg up to 3x a day for children 7 – 12 years
- 500 – 1000 mg up to 3x a day for children > 12 years (and adults)
- Ibuprofen
- (10 mg/kg every 6 hours, max 40 mg/kg/day)
- 200 mg every 6 hours for children 7 – 9 years
- 300 mg every 6 hours for children 10 – 11 years
- 400 mg every 6 hours for children 12 – 17 years
- Not to infants < 6 months
Aspirin is contraindicated due to risk of Reye syndrome, a rare but potentially lethal acute hepatic encephalopathy due to accumulation of salicylate metabolites which can occur in febrile children taking aspirin.
External cooling is not recommended for fever, but it may be used for hyperthermia.
(Febrile seizure)
This is important but it’s not really covered by any topic.
A febrile seizure is an epileptic seizure accompanied by a fever in young children (6 months – 6 years). It affects 3% of children and typically occurs during a benign viral infection. We distinguish simple and complex febrile seizures.
Simple febrile seizures are generalised (symmetric) seizures, last <10 minutes, occurs only once in a 24-hour period, and there are no neurological signs afterward. These are common and harmless.
Complex febrile seizures are focal seizures, last >10 minutes, occurs several times during a 24-hour period, and/or is followed by Todd paresis (transient hemiparesis).
There’s a recurrence rate of 30 – 40%, but there’s no increased risk for developing epilepsy or intellectual disability in case of simple febrile seizures. However, those with complex febrile seizures have an increased risk of subsequent epilepsy.
Rectal diazepam or buccal midazolam can be administered to abort the seizure if it lasts more than 5 minutes, but the febrile seizure usually stops before there is time for admministration in case of first-time seizures. However, for children with history of prolonged febrile seizures, it’s appropriate to provide rescue diazepam or midazolam for the parents to administer the child, should seizures recur. Antipyretics don’t prevent febrile seizures. Antiepileptics are not given to prevent simple febrile seizures, but they may be used in case of complex ones.
There are different definition of overweight and obesity in 2 different topic, see below
“7. Assessment of nutritional status (percentile, BMI, skin fold, laboratory tests). Forms of malnutrition
A child is overweight if their BMI is in the 85th – 95th percentile, and obese if it’s in the 95th percentile or higher.”
“17. Obesity and metabolic syndrome
Overweightness is defined as BMI in the 91st – 98th percentile for age and sex. Obesity is defined as BMI > 98th percentile for age and sex. ”
Which is most correct? Thanks for good notes!
I suspect there are multiple definitions, hence the confusion. I can’t find any definition in the lectures.
The textbook they recommend use the first definition. WHO uses another definition. I don’t know which they prefer, but I’ll change them to be equal at least.
Thank you!
Alkalaemia = pH > 7,45 * 🙂
Hello,
You wrote “PPHN mostly occurs in near-term, term, or post-term infants (not in preterms). ”
But according to Amboss it says “Most commonly term and preterm infants; can also occur in postterm infants”?
When you have two sources with conflicting information, try checking a third source. And then a fourth. And so on.
Other sources agree with me, not Amboss.
My examiner did not like that i mentioned erythema as a symptom of JIA. She says it is a sign of septic arthritis.
Looks like you’re right. Changed