Table of Contents
Page created on December 17, 2018. Last updated on December 18, 2024 at 16:56
Metabolic acidosis
In metabolic acidosis do we have:
- pH below 7.35
- Decreased standard (and actual) serum bicarbonate
- Often a compensatory reduction in pCO2
- Decreased buffer base
- Negative base excess
The word “metabolic” indicates that the primary change that caused this acidosis is a decrease in standard serum bicarbonate. This decreases the [bicarbonate]:[pCO2] ratio (20:1). The lungs will respond by decreasing the pCO2 as well, which increases the ratio toward 20:1 again.
There are two types of metabolic acidosis: Those with normal anion gap and those with elevated anion gap.
The anion gap is a fancy term we use to include all anions in the serum that are not bicarbonate and chloride. The reason they’re called that is because the levels of these anions aren’t measured directly, but rather calculated by the following calculation:
Anion gap = [Na] – ([Cl–] + [HCO3–])
The reason this works is because we know that the plasma is electroneutral, meaning that there must be an equal amount of positive and negative charges. Almost all the positive charge in plasma comes from sodium, while the negative charge comes from chloride, bicarbonate and other anions that aren’t measured in the lab, like lactate, phosphate, sulphate, proteins).
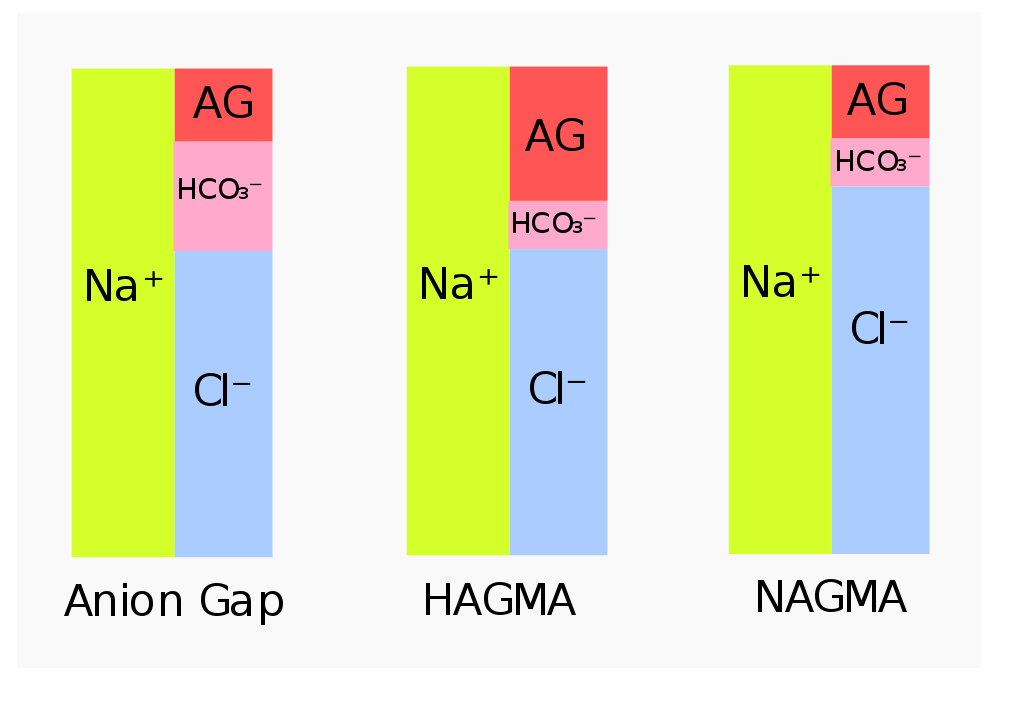
HAGMA = High anion gap metabolic acidosis. NAGMA = Normal anion gap metabolic acidosis. The figure to the left shows the normal situation
The anion gap can be increased due to elevated levels of acids like lactic acid, ketone bodies, ethylene glycol, etc. The extra acids increase the “gap”.
The anion gap is normal in case of increased loss of bicarbonate or decreased loss of H+. No extra acids means that there is no increased “gap”.
Elevated anion gap metabolic acidosis indicates a metabolic acidosis where the [HCO3–] is reduced while the concentration of the anions that comprise the anion gap is increased. It occurs when there is overproduction of organic acids or decreased acid excretion in the kidney.
It can be caused by:
- Renal failure
- Lactic acidosis
- Type A, which is due to increased anaerobic glycolysis
- Type B, which is due to liver disease, so the liver can’t convert lactic acid to pyruvate in the Cori cycle
- Ketoacidosis
- Drug intoxication
- Ethylene glycol (anti-freeze)
- Methanol
In any of these causes will acid accumulate in the serum. In ketoacidosis will there be acidic ketone bodies, in renal failure will H+ excretion be deficient, in lactic acid acidosis will lactic acid accumulate, and so on.
When these acids accumulate in the serum will they donate H+ to the serum, making the serum more acidic and lowering the pH. When the acids have lost this proton will they become anions (A–), meaning that the number of anions in the serum has increased, meaning that the anion gap has increased!
Normal anion gap metabolic acidosis occurs when HCO3– is lost in significant amounts, but no excess acid accumulates in the blood. When bicarbonate is lost will chloride be retained to uphold the electroneutrality. Because of the increased chloride is it sometimes called hyperchloraemic metabolic acidosis. It occurs in two types: Hypokalaemic and hyperkalaemic type. The causes are:
- Hypokalaemic type
- Diarrhoea
- Type I renal tubular acidosis
- Type II renal tubular acidosis
- Carbonic anhydrase inhibitors
In diarrhoea will salt, water, potassium and bicarbonate be lost. Bicarbonate loss causes acidosis, and potassium loss causes hypokalaemia. RAAS activation causes further potassium loss via aldosterone.
In type I renal tubular acidosis are the distal tubules unable to excrete H+, causing acidosis. The bicarbonate regeneration in the distal tubules is also missing, which causes sodium and water loss. The resulting hypovolaemia activates RAAS, causing hypokalaemia.
In type II RTA is there no bicarbonate reabsorption in the proximal tubules. Water and sodium are lost, causing hypovolaemia with RAAS activation.
Carbonic anhydrase inhibitors also disrupt tubular function, causing polyuria with hypovolaemia and RAAS activation.
- Hyperkalaemic type
- Hypoaldosteronism (Addison)
- Type IV renal tubular acidosis
- Potassium-sparing diuretics
Aldosterone stimulates H+ and K+ excretion. In hypoaldosteronism is too little aldosterone produced. In type IV RTA are the tubules resistant against aldosterone. Potassium sparing diuretics work by blocking aldosterone receptors on tubular cells.
Consequences
- Acidosis by itself causes hyperkalaemia. H+ will enter cells in exchange for K+. Hyperkalaemia has cardiac consequences.
- Serum calcium (ionised, Ca2+ form) increases because calcium dissociates from the proteins it’s usually bound to.
- Myocardial contractility decreases.
- Catecholamine sensitivity and sympathetic activation in response to stress is enhanced, causing hypertension.
- Vascular permeability increases.
- Respiratory drive increases (Kussmaul breathing), causing hypocapnia and resulting brain vasoconstriction and hypoperfusion
- Decreased bone mineralization
- Insulin resistance
Compensation
Ventilation will increase, causing a 1.2 mmHg reduction in pCO2 for every 1 mM decrease in bicarbonate.
However, in most cases is the respiratory compensation not sufficient to compensate fully. Recall also that respiratory compensation needs around 12 hours to kick in, so it’s possible that that could be too late for the patient.
Treatment
Treatment is difficult in those cases that include renal problem, so fluid can’t be given. However, when pH drops below 7.1 is sodium bicarbonate infusion necessary.
Hey, Nik!
Under normal anion gap, RTA I, in the last sentence, you mean the hypovolemia is the cause for RAAS activation and not the hypokalemia?
Hey!
Definitely. Fixed. Thanks!