Table of Contents
Page created on October 10, 2018. Last updated on December 7, 2018 at 13:40
Most of the biochemical background of amyloidosis can be read here.
Amyloidosis
Amyloidosis is a disorder of protein misfolding. It results in protein fibrils called amyloids depositing in interstitial space (not inside cells) and cause damage there. The protein fibrils come from proteins that are misfolded, however the specific protein that is misfolded can vary. We know of at least 23 different proteins that can aggregate to form amyloid fibrils.
The reason these proteins aggregate is because when they’re misfolded they expose more β-sheets. These sheets will very easily stack onto each other into a configuration called crossed β-plate. Normally will misfolded proteins be degraded in proteasomes or by macrophages, however in amyloidosis these mechanisms fail for some reason.
Proteins can misfold either because their natural structure gives them a tendency to misfold, or because of a mutation in the protein that make the proteins more prone to misfolding.
The amyloids themselves are not toxic and do not evoke an inflammatory response, but cause damage by compressing healthy cells and tissues until they atrophy due to the pressure.
Types of amyloid
The AL type of amyloid is the most common form. It’s made up of immunoglobin light chains produced by monoclonal plasma cells. AL stands for amyloid light chain. A small minority of patients with AL type amyloidosis have an underlying cancer called multiple myeloma, in which monoclonal plasma cells proliferate uncontrollably. In most AL amyloidosis cases there’s no underlying cancer diagnosis, but they still have some underlying monoclonal plasma cell proliferation that isn’t significant enough to produce a tumour but still significant enough to produce enough light chains to cause amyloidosis.
The AA type of amyloid is made up of a protein called serum amyloid A (SSA), which is an acute phase protein produced by the liver in response to inflammation. It’s usually seen in patients that have chronic inflammation, like in autoimmune diseases like rheumatoid arthritis. AA type is also seen in a febrile disorder called familial Mediterranean fever.
The Aβ type is made up of fragments of a protein called amyloid precursor protein (APP). It’s seen in Alzheimer’s disease.
The ATTR type of amyloid is made up of a protein called transthyretin. This protein is a normal serum protein that carries T4 and retinol. Amyloid formation of this protein is seen in senile systemic amyloidosis.
In haemodialysis-associated amyloidosis the β2-microglobulin forms amyloids in patients that need long-term dialysis.
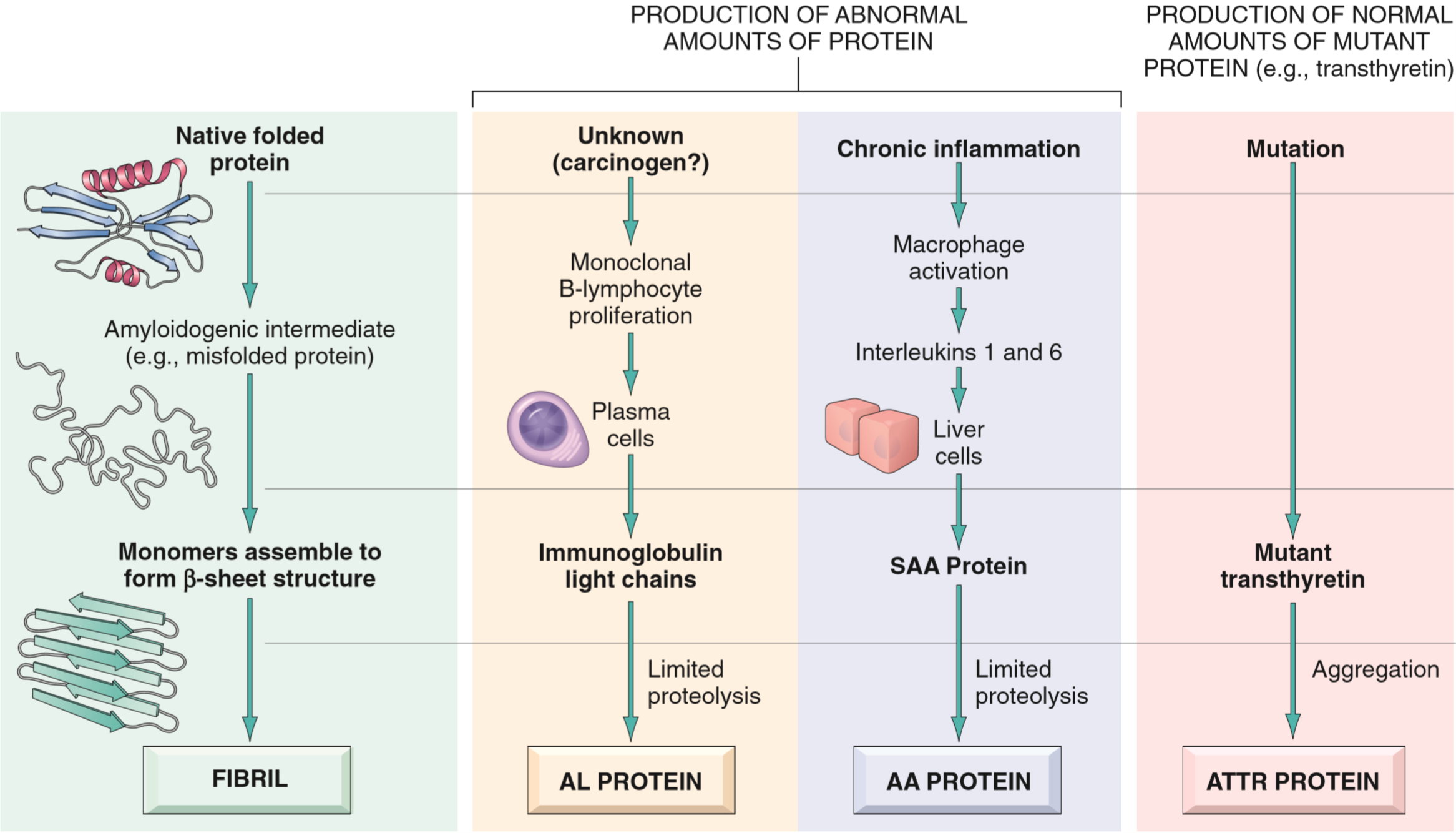
Classification of amyloidosis
The classification of amyloidosis goes like this:
- Systemic (generalized) amyloidosis
- Primary amyloidosis (AL type)
- Secondary amyloidosis (AA type)
- Familial Mediterranean fever (AA type)
- Senile systemic amyloidosis (ATTR type)
- Haemodialysis-associated amyloidosis
- Localized amyloidosis
- Endocrine amyloids in Langerhans islets in type 2 diabetes
- Alzheimer’s disease (Aβ type)
Systemic amyloidosis affects several organ systems. The AA-type is secondary because the amyloidosis is secondary to a chronic inflammation, while the AL-type is primary because all the clinical symptoms come from the amyloidosis itself and not some underlying disease.
Morphology
Organs that are affected by amyloidosis get enlarged and have a greyish discoloration with a waxy, firm consistency.
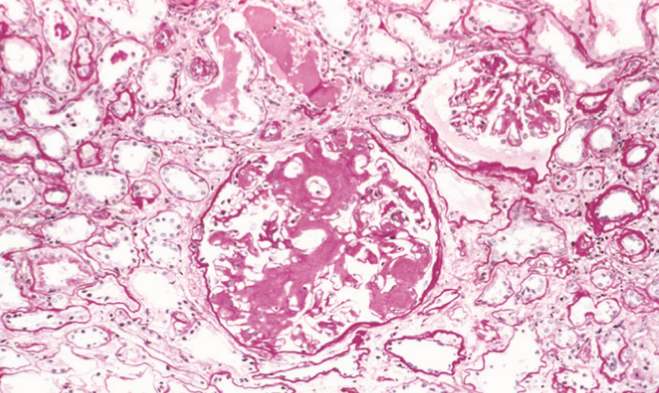
When stained with HE all types of amyloid look bland and hyaline-like. Because of the crossed β-plated configuration of the amyloid fibrils will any type of amyloid stained with Congo red acquire a property called birefringence. This property allows the amyloids to produce a red-green light under a polarizing microscope. The microscopical morphology differs depending on the organ.
In the kidney we can see amyloid deposits in the glomeruli. The deposit begins in the mesangial matrix but progresses to intrude into the capillaries and destroy them.
Amyloidosis of the spleen can have two forms. The “sago spleen” is formed when amyloid only deposits in the splenic follicles. The “lardaceous spleen” form is formed when the amyloid deposits in the splenic sinuses.
Amyloidosis of the liver can be further examined here. The amyloid deposits inside the space of Disse and in sinusoids. The amyloids compress the hepatocytes, causing them to atrophy due to pressure.
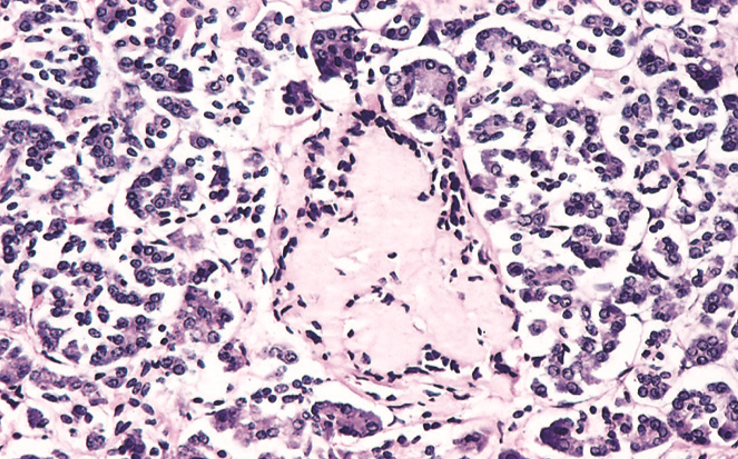
Amyloidosis of the Langerhans islets in the pancreas is seen in long-standing diabetes type 2.
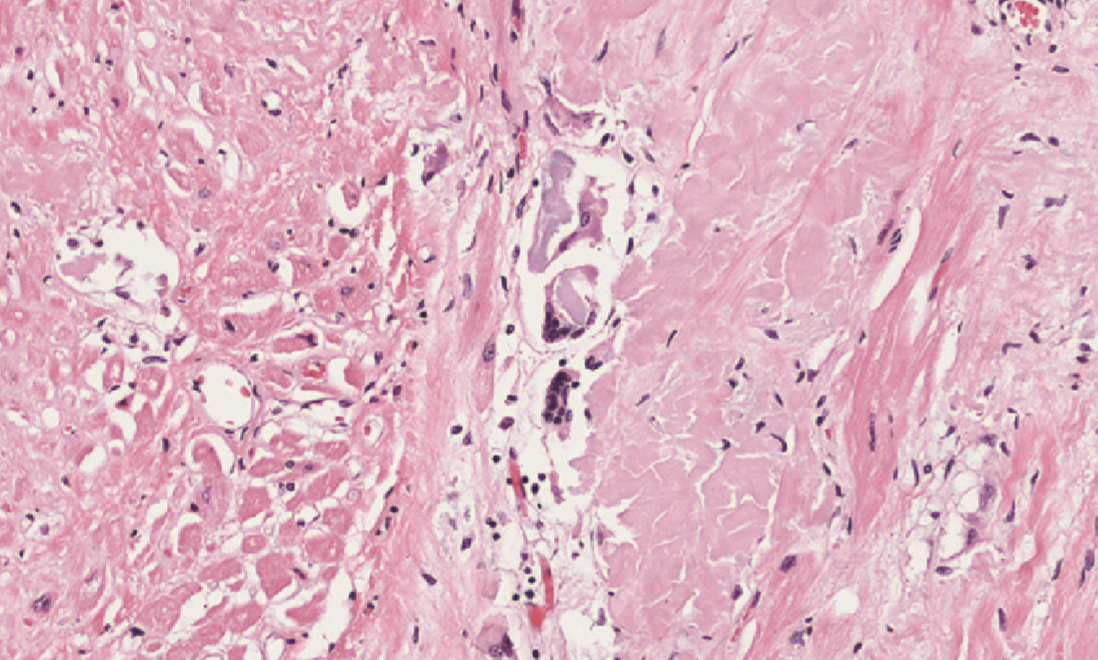
Amyloidosis of the heart causes deposition of amyloids between the myocardial fibres, eventually causing pressure atrophy of them as well.
In systemic amyloidosis the adrenals, thyroid, hypophysis, GI tract and tongue also be affected. The amyloid deposition inside the tongue is important because the it can easily be biopsied to confirm systemic amyloidosis.
Clinical symptoms
Amyloidosis doesn’t always cause symptoms and can sometimes be found during an autopsy. The clinical symptoms depend on the organs affected. They include:
- Weakness
- Fatigue
- Weight loss
- Renal disease
- Proteinuria
- Renal failure is a common cause of death in amyloidosis
- Hepatosplenomegaly
- Cardiac disease
- Conduction disturbances
- Cardiac arrhythmia
The prognosis for systemic amyloidosis is very poor. Mean survival time after diagnosis is 1 – 3 years.
Diagnosis is based on biopsy and Congo red staining of the suspected affected organ. Treatment involves treating the underlying condition if there is one.