Table of Contents
Page created on March 5, 2018. Last updated on December 18, 2024 at 16:55
Learning objectives
- How does fatty acid synthesis inhibit beta oxidation?
- Which factors inhibit fatty acid synthesis?
- Which factors stimulate fatty acid synthesis?
- What are PPARs, and which types exist?
- What activates PPARs in the body?
- Which metabolic processes are controlled by the different types of PPAR?
- How is ketone body synthesis regulated?
- What causes familial hypercholesterolaemia?
- What are statins?
Regulation of fatty acid synthesis
Fatty acid synthesis and beta-oxidation are, like glycolysis and gluconeogenesis, opposite processes. Fatty acid synthesis inhibits beta-oxidation like this. During fatty acid synthesis, acetyl-CoA carboxylase (ACC) produces malonyl-CoA. Malonyl-CoA allosterically inhibits carnitine acyl-transferase I (CAT I), the rate-limiting enzyme of beta-oxidation.
Beta-oxidation doesn’t directly inhibit fatty acid synthesis, but indirectly, as the hormones which stimulate beta oxidation inhibit fatty acid synthesis and vice versa.
Covalent regulation
Acetyl-CoA carboxylase is the rate-limiting enzyme of fatty acid synthesis, and is therefore the point of regulation. ACC is inhibited by PKA and therefore glucagon and epinephrine. It is also inhibited by AMPK. ACC is activated by PP2A and therefore insulin.
Transcriptional regulation
A transcription factor called carbohydrate response element-binding protein (ChREBP) is involved in the transcriptional regulation of fatty acid synthesis. This transcription factor lives in the cytoplasm. AMPK and PKA phosphorylate the transcription factor, which prevents it from entering the nucleus, thereby inhibiting it.
PP2A dephosphorylates the transcription factor. After dephosphorylation ChREBP will travel into the nucleus where it will increase transcription of enzymes like fatty acid synthase, ACC, and pyruvate kinase.
Insulin also stimulates two other transcription factors, SRF and Elk1, which increase the transcription of many genes, including fatty acid synthase, ACC, and pyruvate kinase.
Allosteric regulation
In addition to the factors mentioned above, ACC is also regulated allosterically. Citrate activates ACC as a feed-forward mechanism, while palmitoyl-CoA (palmitate) inhibits it as a negative feedback mechanism.
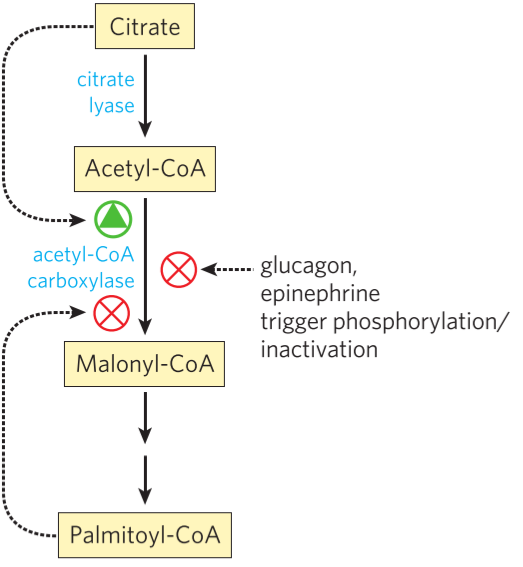
How ACC, and therefore fatty acid synthesis, is regulated. Note that the figure does not include all regulatory factors.
PPAR
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors. This means that they are activated when they bind a ligand. There are 3 types: PPARα, PPARβ/δ, and PPARγ. They are all activated by polyunsaturated fatty acids in the body, especially arachidonic acid.
PPARα turns on genes necessary for uptake and oxidation of fatty acids, which lowers triacylglycerol levels in the blood. Drugs which bind to PPARα (fibrates) are used to treat high triacylglycerol levels in the blood.
PPARβ/δ turns on genes necessary for β-oxidation, and for uncoupling of mitochondria. Uncoupling of mitochondria leads to less effective oxidative phosphorylation, which causes increased energy use and heat production. Activation of these were targets for treating obesity, but early tests show rapid development of cancer.
PPARγ turns on genes necessary for lipid uptake and synthesis. One of the genes is PEPCK. Phosphoenolpyruvate carboxykinase (PEPCK) is an enzyme in the gluconeogenesis, but it is also used for the synthesis of glycerol (glyceroneogenesis). PEPCK is present in adipose tissue for the synthesis of glycerol to be used as a backbone for triacylglycerols. A type of drugs called thiazolidinediones bind to PPARγ and activate it. They could be used to treat diabetes mellitus type 2, but they’re rarely used due to side effects.
Regulation of cholesterol synthesis

The regulation of cholesterol synthesis. The most important are the hormones and AMPK. The transcriptional regulation is not pictured.
Cholesterol cannot be broken down to be used for energy. It is therefore essential that synthesis of it is tightly regulated, and regulated by the body’s availability of energy. All regulation happens on HMG-CoA reductase. The cholesterol levels in the cell can be regulated in five ways:
1. In the short-term, HMG-CoA reductase is regulated covalently by PP2A, PKA, and by AMPK. Insulin activates HMG-CoA reductase through PP2A while glucagon and epinephrine inhibit it through PKA. AMPK also inhibits it.
It’s important to note that the short-term regulation does not depend on the level of cholesterol in the cell, but just on the hormones currently present in the blood. The next four regulation mechanisms depend solely on the level of cholesterol.
2. The long-term regulation happens at the transcriptional level. This means that regulating the number of HMG-CoA reductase molecules present is the most important. If there are too many, transcription of new ones should slow down. If there are too few, transcription should be stimulated.
The transcriptional regulation is accomplished by a pair of proteins called SREBP and SCAP. They are both embedded in the membrane of ER. When the level of cholesterol in the cell drops, they both migrate to Golgi, where SCAP will cleave off a part of SREBP. When that happens, this small part (a regulatory domain) will enter the nucleus and work as a transcription factor to increase transcription of various enzymes, including HMG-CoA reductase.
SREBP = sterol regulatory element-binding protein. SCAP = SREBP cleavage-activating protein
3. Increased levels of cholesterol in the cell activates ACAT. This enzyme attach fatty acids to the cholesterol molecules so that they become cholesteryl esters instead.
4. Increased levels of cholesterol causes the cell to take inn less LDL by endocytosis. LDL contains a lot of cholesterol, so by taking in less LDL the cell won’t accumulate too much cholesterol.
5. Lastly, high levels of cholesterol in the cell causes the cell to remove molecules of HMG-CoA reductase by proteolysis. By reducing the number of this enzyme cholesterol synthesis will be slowed down.
Regulation of ketone body synthesis
When the liver has run out of both glycogen stores and gluconeogenesis substrates, fats will be broken down for energy. Beta-oxidation will yield acetyl-CoA from fatty acids.
Recall that oxaloacetate is a gluconeogenetic substrate. In this scenario the liver has run out of easily available gluconeogenetic substrates, including oxaloacetate. Oxaloacetate is necessary to metabolize acetyl-CoA through the TCA cycle. Because there is no oxaloacetate, acetyl-CoA will accumulate. It is the accumulation of acetyl-CoA which stimulates ketone body synthesis, as the acetyl-CoA has nowhere else to go.
To maintain blood sugar at a minimal level the body will start to break down proteins for glucogenic amino acids, and use them as substrates for gluconeogenesis. However, these are not sufficient to provide energy to all organs.
Hormones also influence ketone body synthesis. The gene for HMG-CoA synthase is regulated by a transcription factor called FOXA2. Insulin inhibits this transcription factor, thereby inhibiting ketone body synthesis. Glucagon activates it, thereby stimulating ketone body synthesis.
A protein called SIRT3 activates HMG-CoA synthase covalently by deacetylating it. SIRT3 is activated by fasting.
Disorders
Hypercholesterolaemia
Hypercholesterolaemia is a condition where you have increased level of cholesterol and LDL in the blood. It leads to atherosclerosis and cardiovascular disease, including myocardial infarction and stroke.
Most cases of hypercholesterolaemia is due to poor diet and inactivity. However, there are some genetic causes. Familial hypercholesterolaemia is due to defective LDL receptors. With these receptors defective cells can’t take in LDL, causing it to accumulate in the blood. Cholesterol accumulates in the skin, and early atherosclerosis causes cardiovascular diseases to occur in a very early age.
Hypercholesterolaemia is generally treated with exercise and improved diet. If unsuccessful, drugs can be used. One of the most important drugs in treating cardiovascular disease are the statins. These drugs inhibit HMG-CoA reductase, reducing the synthtesis of cholesterol.
Disorders of fatty acid oxidation
Deficiency of acyl-CoA dehydrogenase or primary carnitine deficiency decreases the formation of ketone bodies. Fat accumulates in the tissues, causing toxic damage.
Acidotic disorders
These disorders cause acidosis, the decrease of blood pH. Methylmalonic acidaemia is caused by a genetic defect in the B12-dependent enzyme methylmalonyl-CoA mutase. Propionic acidaemia is caused by a genetic defect in the biotin-dependent enzyme propionyl-CoA carboxylase. These diseases cause a build-up of organic acids in the blood.
Sphingolipidoses
Sphingolipidoses are a type of lysosomal storage diseases caused by abnormal sphingolipid metabolism. They include Tay-Sachs disease, Fabry disease, Gaucher disease, Niemann-Pick disease, and Krabbe disease.
Summary
- How does fatty acid synthesis inhibit beta oxidation?
- The product of ACC, malonyl-CoA, inhibits carnitine acyl-transferase I
- Which factors inhibit fatty acid synthesis?
- Phosphorylation by PKA, allosterically by palmitate
- Which factors stimulate fatty acid synthesis?
- Dephosphorylation by PP2A, increased transcription by PP2A, allosterically by citrate
- What are PPARs, and which types exist?
- PPARs are ligand-activated transcription factors
- PPARα, PPARβ/δ, and PPARγ exist
- What activates PPARs in the body?
- Polyunsaturated fatty acids, especially arachidonic acid
- Which metabolic processes are controlled by the different types of PPAR?
- PPARα turns on genes necessary for uptake and oxidation of fatty acids
- PPARβ/δ turns on genes necessary for β-oxidation, and for uncoupling of mitochondria
- PPARγ turns on genes necessary for lipid uptake and synthesis.
- How is ketone body synthesis regulated?
- In a fasting state the body has run out of easily available gluconeogenetic substrates, including oxaloacetate. Beta-oxidation of fatty acids will yield acetyl-CoA. Without oxaloacetate acetyl-CoA can’t be broken down in the TCA cycle, so they will be shunted towards ketone body synthesis
- A protein called SIRT3 is activated by fasting. It deacetylates and therefore stimulated HMG-CoA synthase.
- What causes familial hypercholesterolaemia?
- Defective LDL receptor
- What are statins?
- Statins are drugs which inhibit HMG-CoA reductase
- They’re used to treat hypercholesterolaemia