Table of Contents
Page created on March 7, 2018. Last updated on December 18, 2024 at 16:56
Summary
- Hemoglobin is comprised of four chains, 2 alpha and 2 beta chains, and four heme units
- Sickle cell disease occurs after a mutation in the gene for the beta chain
- Problems with the gene for the α-chain causes α-thalassemia, while too little β chain cause β-thalassemia
- HbA1c is hemoglobin glycated with glucose on the β-globin chain and its level shows the long term average blood glucose level
- Every person has 4 genes coding for the alpha chain and 2 genes coding for the beta chain
Sickle cell disease
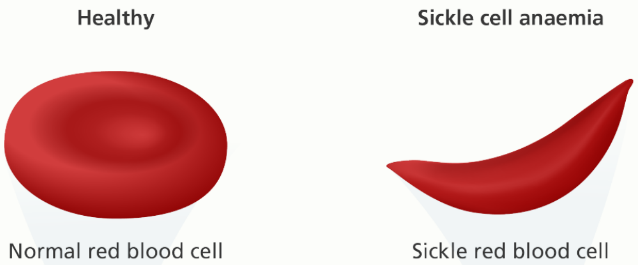
Sickle cell disease is a disease where a mutation in one or both of the genes for the beta chains causes the RBCs to take on a sickle-shaped form, which reduces their ability to circulate. The mutation in question is a substitution of valine for glutamate at the sixth position from the N-terminal end of the β-chain of the hemoglobin. This mutation is formulated like this: α2β26Glu-Val. This causes a shift from the normal hemoglobin type “A” to type “S”.
We carry two copies of each gene, and to have sickle cell disease, both copies of the beta chain gene must have the S mutation. Sickle cell disease is therefore often denoted Hb SS. When deoxygenated, Hb S polymerises to form the characteristic sickle-shape, which lacks the normal flexibily of RBC and therefore causes problems.
When a person is heterozygous for this mutation (carries one mutated and one normal gene), they are carriers for sickle cell disease and have a condition called sickle cell trait. People with sickle cell trait are also resistant to malaria infection. When malaria virus infects RBCs of someone with this trait, the virus will cause oxidative stress in the cell. However, the red blood cells of people with sickle cell trait are very vulnerable to oxidative stress, so when they are infected by the malaria virus the RBC will immediately change into the sickle-shape. The sickle-shaped RBC is then eaten by phagocytes, so the virus can’t spread. Because of natural selection, the sickle cell trait is prevalent in places where malaria is also prevalent.
Hemoglobin C
Hb type C is found almost exclusively in black populations. It forms as a result of a similar mutation as in Hb SS, however glutamate is substituted for lysine instead of valine. Therefore, it’s denoted α2β26Glu-Lys. They are mostly asymptomatic, with the exception of mild chronic hemolytic anaemia and formation of Hb C crystals in the cells.
α-thalassemia
Unlike for the beta chain are there 4 genes in each cell that code for the α-chain. A problem with two or more of these genes will manifest as a type of α-thalassemia. Normal people have αα/αα (all 4 genes intact), while silent carriers of this disease have -α/αα (one gene deleted). People with either -α/-α or –/αα (two genes deleted) have α-thalassemia minor. People with –/-α have only one copy of the gene, which is called Hb H disease. Having no functional copies of the gene (–/–) is called Bart’s hydrops fetalis syndrome and is incompatible with life. The fetus will die in utero or shortly after birth.
| Normal | αα/αα |
| Silent carrier | -α/αα |
| Minor | -α/-α or –/αα |
| Hemoglobin H disease | –/-α |
| Barts hydrops fetalis | –/– |
People with α-thalassemia minor have mild microcytic hypochromic anaemia. People with Hb H disease are worse off. They live normal lives until an infection, pregnancy or exposure to oxidative drugs trigger a hemolytic crisis, which causes acute and severe anaemia.
β-thalassemia
The β-types of thalassemias are more serious than alpha-type. In this disease, something is wrong with β-globin synthesis due to mutations in its gene. Often, the rate of synthesis is insufficient. Like in α-thalassemia, having one defect and one working gene gives you β thalassemia minor. It usually results in mild asymptomatic hemolytic anemia and requires no treatment usually.
β-thalassemia intermedia falls between minor and major. Patients with this disease may require blood transfusion regularly.
The major type of the disease is obviously the most severe. It is characterized by very severe microcytic hypochromic anemia, with very low levels of Hb in the blood. This causes characteristic changes in the bones, as the body tries to increase the amount of bone marrow to increase erythropoiesis. They also require regular transfusions, and iron chelation to remove excess iron. It notably causes splenomegaly. The only cure for this disease is bone marrow transplantation.