Page created on December 12, 2018. Last updated on December 18, 2024 at 16:56
Coagulopathies
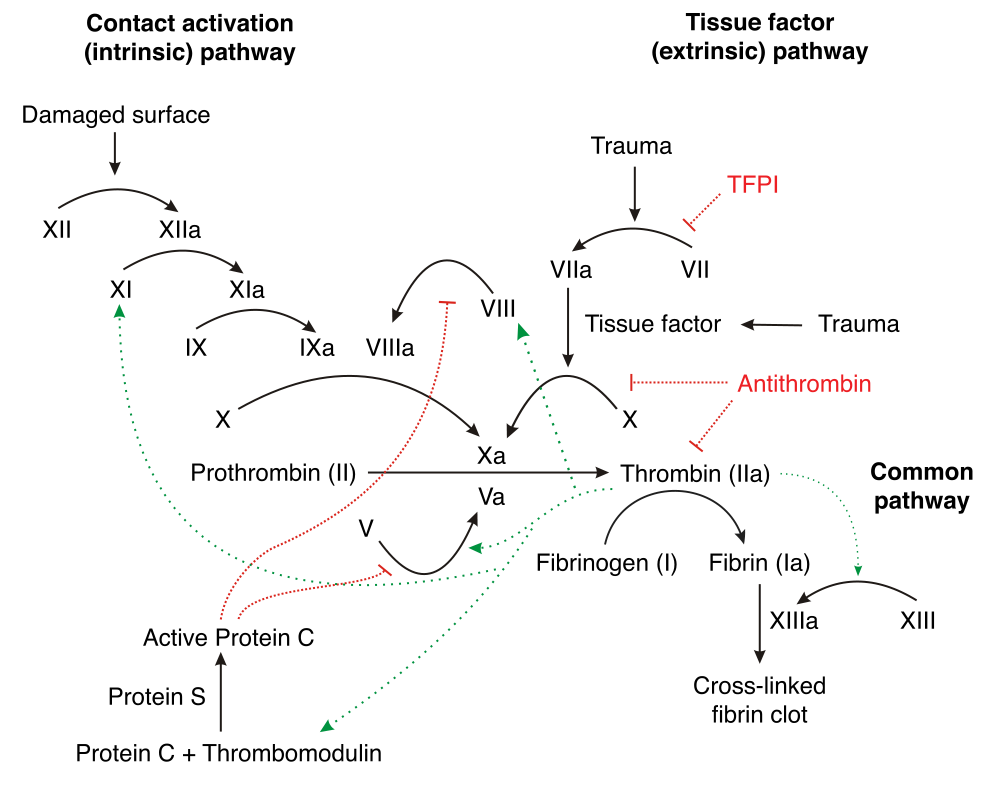
Disorders where problems with the clotting factors in the blood are called coagulopathies. The goal of the clotting cascade is to form fibrin from fibrinogen. While problems with the vessels or platelets cause small bleedings will coagulopathies cause large bleedings, like haematomas. The bleeding time is normal (except in von Willebrand disease), but the clotting time is prolonged.
The classification goes like this:
- Hereditary
- Haemophilia A – factor VIII deficiency
- Haemophilia B – factor IX deficiency
- Haemophilia C – factor XI deficiency
- von Willebrand disease – von Willebrand factor deficiency
- Acquired
- Vitamin K deficiency
- Liver failure
- Antibodies against factor VIII
- Drug overdose
- DIC
Haemophilia A is an X-linked recessive disease, meaning that it mostly affects males. Factor VIII is important in the intrinsic coagulation pathway. The severity of bleeding depends on the degree of factor deficiency. In the most severe cases, huge bleedings occur, like haematomas, haemarthros and retroperitoneal bleedings cause arthrosis, muscular and neural damage. The treatment is replacement of factor VIII by giving fresh blood or plasma. A potential complication is the formation of anti-factor VIII antibodies. Many transfusions carry the risk of HIV and hepatitis infections.
Haemophilia B is also X-linked recessive. Factor IX is also important in the intrinsic pathway. The symptoms are similar to those of haemophilia A. The treatment is replacement of factor IX by transfusion, which carries the same risk as for haemophilia A.
Haemophilia C is rare and mild, because factor XI is only involved in the intrinsic pathway, leaving the extrinsic pathway perfectly fine and able to coagulate the blood.
von Willebrand disease is an autosomal dominant disease, so the genders are more equally affected. von Willebrand factor binds to factor VIII in the plasma and keeps it stable. It’s also important in promoting platelet adhesion. When vWF is deficient will factor VIII be deficient as well, because without vWF is factor VIII rapidly degraded. Heterozygotes have the mild form, where they only bleed severely during trauma or surgery. Special for von Willebrand disease is that both clotting and bleeding time are prolonged.
Vitamin K is needed for the posttranslational modification of proteins that need to bind calcium, including factor II, VII, IX, X and proteins C and S. When vitamin K is deficient will these factors not be produced.
Liver failure impairs the liver’s ability to synthesize the clotting factors I, II, V, VII, IX, X, XI and proteins C and S and antithrombin III. Liver failure also leads to portal hypertension, which leads to splenomegaly, which leads to increased sequestration of platelets and RBCs.
Anti-factor VIII antibodies can occur in long-term treatment with factor VIII in haemophilia A, in SLE, in elder people or after giving birth.
Heparin inhibits factor II (thrombin) while streptokinase activates fibrinolysis. They’re both used for treating acute myocardial infarcts.
And for Haemophilia A as well, shouldn’t it only affect the intrinsic pathway?
True, both factors are in the intrinsic pathway. The book writes that VIII is involved in the common pathway, which I don’t think is right.
Nice catch!
Shouldn’t Haemophilia B affect only the intrinsic pathway?