Page created on May 17, 2019. Last updated on December 18, 2024 at 16:57
General
Adrenogenital syndrome is an outdated expression; nowadays congenital adrenal hyperplasia (CAH) is preferred.
Congenital adrenal hyperplasia is a group of autosomal recessive diseases characterised by adrenal hyperplasia. It’s caused by enzyme defects of some of the enzymes that are involved in hormone synthesis in the adrenal cortex.
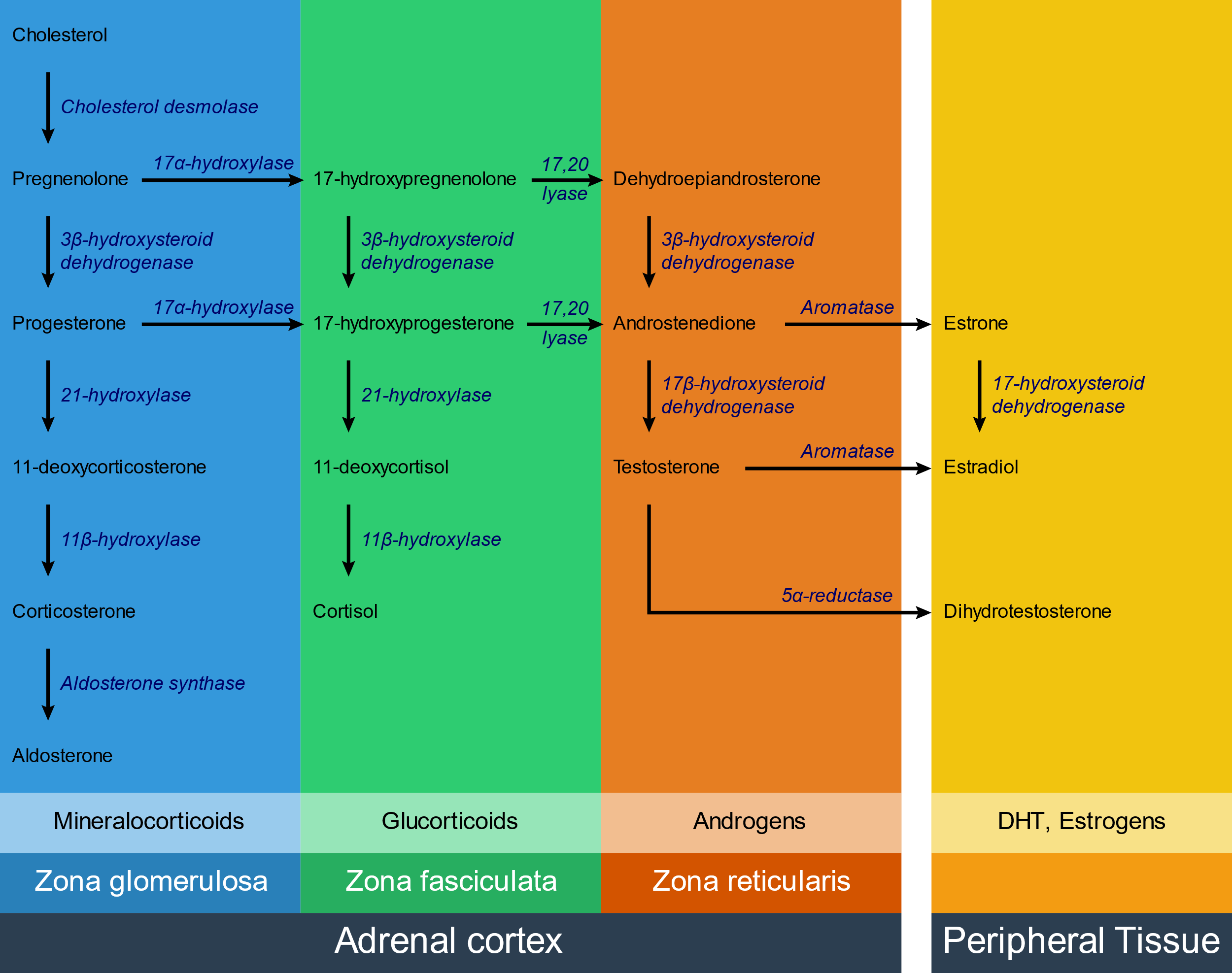
The exact clinical manifestations depend on which enzyme is defective, but all types are characterised by low levels of cortisol, high levels of ACTH and adrenal hyperplasia. In some types the level of androgens is increased, which causes virilization (masculinization) of the female genotype.
Etiology:
The following enzymes can be affected:
- 21-hydroxylase – 95% of cases
- 11β-hydroxylase – 5% of cases
- 17α-hydroxylase – rare
The most commonly affected enzyme is 21-hydroxylase.
Pathomechanism:
The enzyme defects are rarely absolute, meaning that the enzyme function is rarely 100% lost. There are two main possibilities:
- Most of the enzyme function is lost – symptoms are seen right after birth
- Only some of the enzyme function is lost – symptoms are seen in adolescence
When one enzyme is defective the substrate for that enzyme will accumulate right before the enzyme block. In any of the aforementioned enzyme defects, cortisol will be deficient. This decreases the negative feedback on ACTH, causing it to rise. Increased ACTH causes adrenal hyperplasia and increases production of steroid hormone precursors. These precursors will then travel down the functioning pathways, which is often just the androgen pathway.
The following table shows how hormone levels change in the various enzyme defects:
| Enzyme | Cortisol | Aldosterone | 11-Deoxocorticosterone (DOC) | Androgens |
|
21-hydroxylase |
↓ | ↓ | ↓ | ↑ |
| 11β-hydroxylase |
↑ |
|||
| 17α-hydroxylase |
↓ |
DOC is a weak mineralocorticoid; however due to the adrenal hyperplasia it is produced in very large amounts, which can compensate for the lack of aldosterone and even cause hypertension.
Clinical features:
| Enzyme | Blood pressure | XX genotype (female) | XY genotype (male) |
| 21-hydroxylase | Hypotension | Clitoromegaly/presence of male genitalia. Virilization. | Normal male genitalia. Early puberty. |
| 11β-hydroxylase | Hypertension | ||
| 17α-hydroxylase | Normal female genitalia at birth. Delayed puberty. | Female external genitalia at birth. Delayed puberty. |
Virilization involves developing male characteristics like deep voice and increased body hair.
Some symptoms are common for all types:
- Hypoglycaemia – due to decreased cortisol
- Hyperpigmentation – as melanocyte-stimulating hormone (MSH) is produced together with ACTH
Complications:
Due to the cortisol deficiency these patients are at risk for adrenal crisis, an acute severe glucocorticoid deficiency that can be triggered by stress or other factors. Fever and vomiting develops and patients are at risk for developing severe shock.
Treatment:
Treatment involves substituting the deficient hormones and blocking the excess hormones. All types are treated with glucocorticoids. 11β and 17α are treated with mineralocorticoid receptor blockers as well (spironolactone). 21 also needs mineralocorticoid substitution.
Hypogonadism
Hypogonadism refers to impaired functional activity of the gonads. We distinguish two types:
- Primary hypogonadism (hypergonadotropic hypogonadism) – where the steroid production in the gonads is deficient
- Secondary hypogonadism (hypogonadotropic hypogonadism) – where the GnRH production in the hypothalamus or FSH, LH production in the pituitary is deficient
Etiology:
Both genetic defects, chromosomal aberrations and acquired etiologies exist.
- Primary hypogonadism
- Turner syndrome (45X0)
- Klinefelter syndrome (47XXY)
- Androgen insensitivity syndrome
- Anorchia
- Trauma
- Chemotherapy
- Irradiation
- Autoimmune disease
- Infections
- Mumps
- Tuberculosis
- Secondary hypogonadism
- Kallmann syndrome
- Pituitary neoplasms
- Trauma
Pathomechanism:
In primary hypogonadism the decreased gonadal steroid production causes FSH and LH to increase by decreased negative feedback.
In secondary hypogonadism the decreased level of FSH and LH causes decreased gonadal steroid production.
The symptoms depend on when the hypogonadism occurs. If it is congenital males can develop female-like genitals.
Clinical features:
The clinical features are defined by the lack of sex hormones. The symptoms are different depending on whether hypogonadism occurs before or after puberty. Puberty is often delayed.
Males:
- Prepubertal
- Long extremities (delayed closure of epiphyseal plates)
- Little body hair
- Poor development of skeletal muscles
- Abnormal genitalia
- Undescended testes
- Hypospadias
- Postpubertal
- Decreased sexual function
- Lack of energy
Females:
- Prepubertal
- Primary menorrhoea – no menses before the age of 16
- Atrophic genital organs
- Postpubertal
- Secondary amenorrhoea – irregular menses in women who previously had normal menses
Hypergonadism
Hypergonadism is much less frequent than hypogonadism. It’s often due to sex-hormone producing tumors in the gonads.
Etiology:
- Gonadal hormone-producing tumors
- Anabolic steroid abuse
- Liver disease (decreased clearance of sex hormones)
Clinical features:
Hypergonadism causes precocious puberty, where a person enters puberty earlier than normal. It’s defined as onset of puberty before:
- Age 8 in females
- Age 9 in males
I meant the other way around with the enzymes.
Then yes, 17-hydroxylase deficiency should increase aldosterone levels, however the large amounts of DOC causes a significant negative feedback on the aldosterone secretion, ultimately causing its levels to be decreased.
I never stated that 11-hydroxylase deficiency cause males to develop female genitalia. Those with 17-hydroxylase deficiency do, however.
Szia, Nik
In 17 hydroxylase def. you will have a large amount of aldosterone(blue book, p.368) DOC will not be decreased.
You also state in your second table that XY will develop female genetalia and early puberty in 11beta-hydroxylase def.
Always question the blue book. Aldosterone will be low. Check instead virtually any other source, like these: https://emedicine.medscape.com/article/920532-workup
and here:
https://www.uptodate.com/contents/uncommon-congenital-adrenal-hyperplasias#H1698113
But yeah, the “normal female genitalia” was a typo. It’s fixed now.
I think that aldosterone will increase in 11Beta hydroxylase defiency? And also in the 17 hydroxylase there will be increase in androgen production of male which can cause early puberty and negative feedback to the testes causing testicular atrophy, but they won’t develop female genitalia
Do you have a source? Everything you stated contradicts my sources, amboss and emedicine.