Table of Contents
Page created on January 7, 2019. Last updated on December 18, 2024 at 16:57
Summary
- The body metabolizes drugs in two steps; phase I and phase II
- Phase I, where the drug is oxidized, reduced or hydrolysed to yield an -OH group
- The most important enzymes in phase I reactions are the CYP450 enzymes, monoamine oxidase, alcohol dehydrogenase, etc
- Phase II, where the enzyme attaches a chemical group to the -OH group on the drug
- The important enzymes here are transferases, enzymes which conjugate the drug with a chemical group
- Commonly conjugated chemical groups are glucuronic acid, sulphuric acid, glycine, glutathione, acetyl and methyl
- Biotransformation increases water solubility of the drug, making it easier to excrete, and it (most frequently) inactivates the drug.
- There are 4 possible outcomes of a biotransformation reaction:
- The active parent drug is converted into an inactive metabolite (most common)
- The inactive parent drug is converted into an active metabolite. This is what occurs when you give a prodrug
- The active parent drug is converted into an active metabolite
- The parent drug is converted into a toxic metabolite
- Biotransformation of paracetamol produces a toxic metabolite called NAPBQI. In paracetamol overdose this metabolite accumulates, causing liver necrosis
- Treatment for this is acetylcysteine
- Neonates have decreased UGT activity, decreasing their elimination of chloramphenicol
- Neonates have decreased CYP450 activity. CYP450 and UGT metabolizes diazepam, so they have decreased elimination of it
- Neonates have decreased ADH activity, reducing their elimination of alcohol
- There are large genetic differences in biotransformation (pharmacogenetics)
- Many Scandinavians are so-called slow acetylators and have reduced elimination of drugs which must be acetylated, like isoniazid
- CYP450 enzymes can be induced or inhibited
- Induction is usually due to drugs binding to a receptor which increases the transcription of the enzyme
- Inhibition is usually competitive
- Inhibition can be exploited clinically
- Ritonavir is a CYP3A4 inhibitor, and is often given with other antiretrovirals which are metabolized by CYP3A4
- Ketoconazole is also a CYP3A4 inhibitor, and is often given with cyclosporin, which is metabolized by CYP3A4
Introduction
There are three ways drugs can be eliminated from the body:
- The drug can be biotransformed into an inactive metabolite, which is then excreted via feces or urine
- The drug is excreted without being biotransformed
- Some of the drug is biotransformed, so both the unchanged drug and inactive metabolites are excreted
We’ve already encountered biotransformation in biochemistry 2. During biotransformation a drug is converted into one or more metabolites by enzymes. These metabolites may undergo further biotransformation.
The drug that was administered and which has not been biotransformed yet is called the parent compound (because it is the “parent” of the metabolites).
Biotransformation predominantly occurs in the liver, but also in the intestinal mucosa, renal tubular cells and in the colon by bacteria. We’ll mostly cover the liver here.
Biotransformation
The ultimate goal of biotransformation is to attach a chemical group to the drug molecule. By doing this will the biological activity of the drug be inhibited, and it becomes more water-soluble, which makes it easier to excrete. However, the liver can’t just attach any chemical group to any drug molecule. That’s not how chemistry works.
For the liver to attach a chemical group to the drug molecule it must first create an -OH group on the molecule that it can attach the chemical group to. This is the phase I reaction of biotransformation.
Now that the drug has an -OH group the liver will attach a chemical group to the -OH group. These chemical groups can be glucuronic acid, sulfuric acid, glycine and so on. The drug is now no longer the same as it was when it was administered – it is now a metabolite.
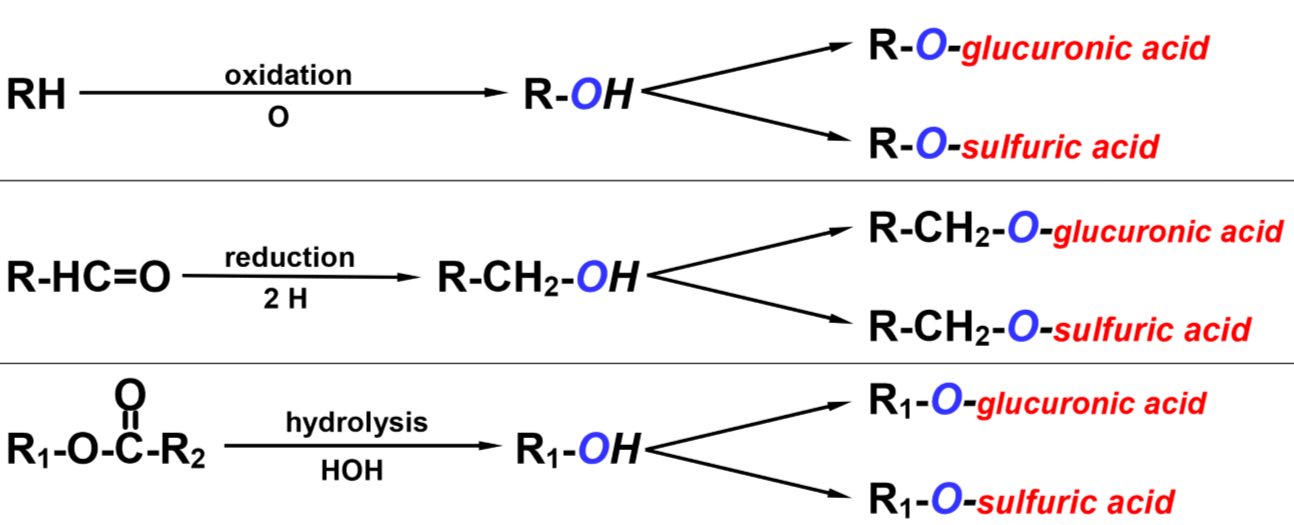
Typical mechanisms of biotransformation. The “R” here signifies the drug molecule itself.
Let’s look at these two in more detail.
Phase I biotransformation
For the liver to create an -OH group on the drug the liver must perform one of these three reactions on the drug:
- Oxidation (most frequent)
- Reduction
- Hydrolysis
The drug now went from being R-H or something similar to becoming R-OH. It is now a phase I metabolite, which has slightly higher water-solubility but typically the same biological activity, so the liver must still perform phase II.
Note that other groups than -OH can be used as well, such as -NH2, -SH and -COOH, however I’ll continue to use -OH as an example because it’s the most frequent type.
Oxidation:
The cytochrome P450 superfamily is essential for phase I. CYP450 is a superfamily of enzymes that mostly catalyse oxidation but can perform dehydrogenation and reduction as well. CYP450 is found both in the liver and in the intestinal mucosa. The superfamily is classified into families, which are denoted by number, which are further divided into subfamilies, denoted by capital letters, which contains the individual enzymes, which again are numbered.
So, CYP3A4 is the 4th enzyme of the A subfamily of the 3rd family of CYP450 enzymes. CYP3A4 is also the most abundant CYP enzyme, both in the liver and the intestinal mucosa.
Why are CYP450 enzymes in the intestinal mucosa? Because the intestinal mucosa is the first organ any ingested drug or toxic substance meets. For the body, the earlier biotransformation begins, the better.
Flavin-containing monooxygenase (FMO) is another ER-bound enzyme involved in phase I. It catalyses monooxygenation like the name suggests, where 1 atom from O2 is inserted into the drug to give an -OH group. Like the name suggests does it contain a flavin-group, which comes from riboflavin, vitamin B2.
Some other enzymes that catalyse oxidation aren’t found in the ER, so they’re non-microsomal. They are:
- Monoamine oxidase (MAO) – in the mitochondria
- Aldehyde oxidase (AOX) – in the cytosol
- Xanthine oxidase (XO) – in the cytosol
- Alcohol dehydrogenase (ADH) – in the cytosol
- Aldehyde dehydrogenase (ALDH) – in the cytosol
Reduction:
Reduction is catalysed by different enzymes, like:
- Microbial enzymes produced by bacteria in the colon
- CYP450
- Aldo-keto reductase (AKR)
- Carbonyl reductase (CBR)
- Methionine sulfoxide reductase (MSR)
Hydrolysis:
Hydrolysis is catalysed by other enzymes however I’m not going to list them as I don’t believe it’s important.
Phase II biotransformation
Now that the drug has an -OH group other enzymes can catalyse the conjugation (attachment) of certain chemical groups to this -OH group, which will make the drug more water soluble and less biologically active.
The enzymes that catalyse phase II are called transferases, because they transfer chemical groups from endogenous cosubstrates onto the drugs. What chemical groups can be transferred? These are the most common:
- Glucuronic acid
- Sulphuric acid
- Glycine
- Glutathione
- Acetyl
- Methyl
Which chemical group is transferred depends on the drug. For example is morphine always glucuronidated, L-DOPA always methylated and minoxidil always sulphated.
The most important phase II enzymes are UDP-glucuronosyl
transferases (UGT) and N-acetyltransferases (NAT).
Now that the drug has been conjugated with a chemical group is it now:
- More water soluble, making it easier to excrete
- Less biologically active (in most cases)
Why only in most cases?
Influence of biotransformation on the biological activity of drugs
There are four possible outcomes of biotransformation of a drug molecule:
- The active parent drug is converted into an inactive metabolite (most common)
- The inactive parent drug is converted into an active metabolite. This is what occurs when you give a prodrug
- The active parent drug is converted into an active metabolite
- The parent drug is converted into a toxic metabolite
Scenario 1 is the most typical. After conjugation the drug becomes inactive, which is what the body tries to achieve by biotransformation. This is what occurs with propranolol, theophylline, paracetamol and so on.
Scenario 2 is what occurs when you give a prodrug. A prodrug is a drug that isn’t active in the body by itself. However, after being biotransformed it will become an active metabolite which has effect on the body.
In some cases it turned out that certain drugs that had been available for a long time were actually prodrugs. In other cases are drugs specifically designed to be prodrugs. If the active drug has low oral bioavailability or low water solubility for example can we sometimes produce a prodrug that don’t have these drawbacks, and the body will convert the prodrug into the active drug by itself.
Scenario 3 is rarer. In these cases are not only the parent drugs active, but their metabolites also, which slows down the elimination of the drug. This includes drugs like codeine (which is converted to morphine by the liver, which is also active), diazepam and morphine itself. So codeine is converted into morphine which is converted into morphine 6-glucuronide, and all three are active!
Scenario 4 is when a drug is converted into a toxic metabolite. This is usually not a problem, because the body can usually deal with the toxic metabolite before it accumulates. However, this has consequences in overdosing. Let’s look at an example.
Paracetamol can be bought without a prescription, so it’s not thought of as dangerous. Paracetamol in the liver can be biotransformed by two pathways. It can either:
- Be dehydrogenated by CYP2E1 into a toxic metabolite called NAPBQI
- Be glucuronidated by UGT into paracetamol-glucuronidate.
Paracetamol-glucuronidate isn’t toxic and is excreted without problem. NAPBQI however, is toxic, as it binds to proteins and enzymes and inactivates them. In a healthy liver will some amounts of both metabolites be produced, however NAPBQI is quickly detoxified by conjugation with glutathione, forming a non-toxic metabolite that is excreted without any problem.
If someone overdoses on paracetamol (which is 12g for healthy people) will glutathione stores in the liver be consumed and depleted, so that the liver can no longer detoxify NAPBQI, causing it to accumulate and cause liver necrosis!
This is also significant for alcoholics, for two reasons: The CYP2E1 enzyme is upregulated in alcoholics, because CYP2E1 also inactivates ethanol. Also, the livers of alcoholics have decreased amount of glutathione
Because they have more CYP2E1 enzyme more paracetamol is converted into NAPBQI and less is inactivated by glucuronidation. Also, because they have less glutathione they run out of it more quickly. These factors result in that chronic alcoholics can only tolerate 4-5 grams of paracetamol, which is almost 1/3 of what healthy people can tolerate!
The antidote for paracetamol overdose is N-acetylcysteine, because the liver converts it to cysteine which is an important part of glutathione. This replenishes the glutathione stores.
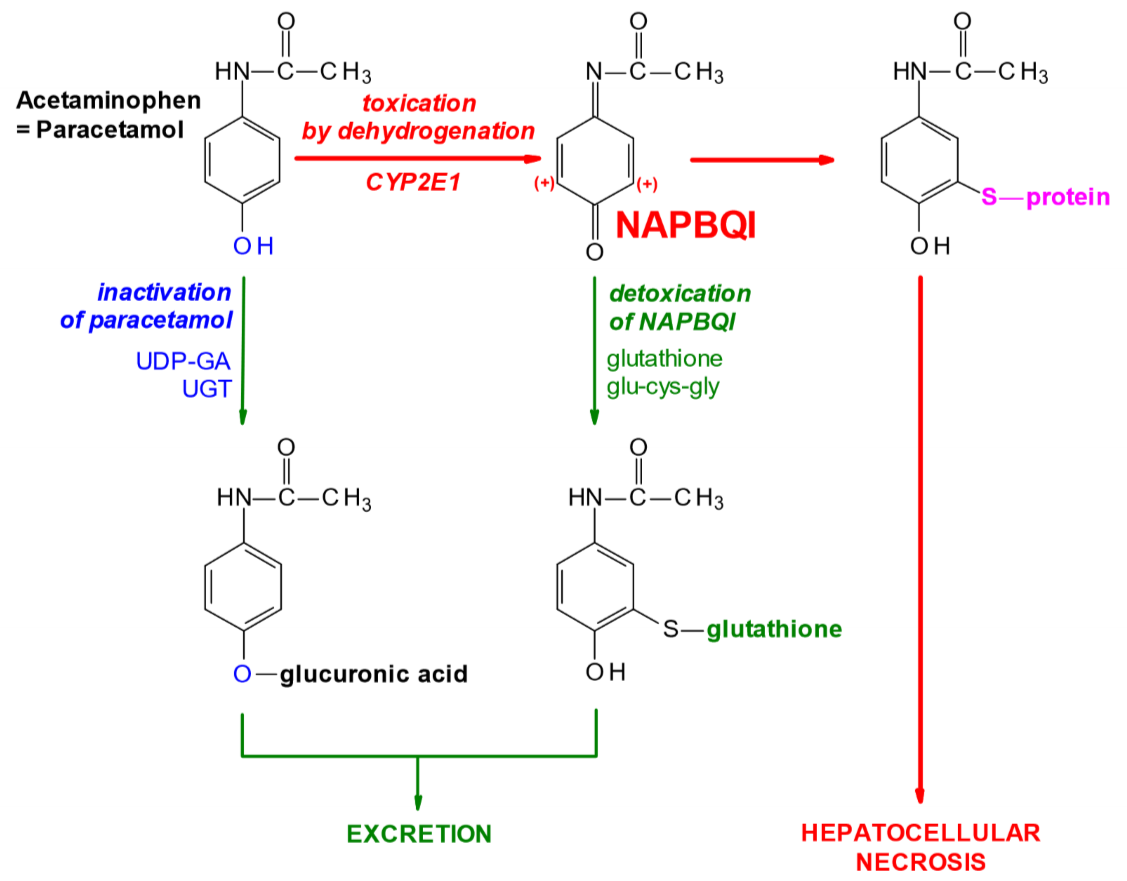
Biotransformation of paracetamol (=acetaminophen)
Alterations in biotransformation
In neonates:
Neonates are unfortunate. Their biotransformation enzymes aren’t fully mature and active yet, so they are prone to some unfortunate conditions. Here are some examples:
- The enzyme that conjugates bilirubin, UGT, is under-expressed in neonates. This means that they are prone to physiological jaundice because the body can’t excrete bilirubin normally yet. This is harmless.
- UGT also inactivates chloramphenicol. This is the reason chloramphenicol can’t be used in neonates, because the impaired biotransformation of it will cause grey baby syndrome
- In addition to under-expressed UGT don’t neonates have fully expressed CYP450 enzymes either. Both enzymes are needed to biotransform diazepam. The inability to get rid of diazepam is what causes floppy infant syndrome in babies whose mother received diazepam before delivery
- Neonates don’t have much alcohol dehydrogenase (ADH) either. If the mother had ethanol in their system before the delivery will the baby be born with significant blood ethanol concentration. Neonates need a lot of time to get rid of this ethanol with the low amounts of ADH they have.
Genetics:
Genetics also play a role here. Genetic mutations in biotransformation enzymes can cause people to biotransform certain drugs either too slowly or too quickly. This is called pharmacogenetics and has its own topic in pharma 3. Let’s look at some examples.
- > 70% of Scandinavians and middle eastern people have deficiency of an enzyme called NAT2. This enzyme acetylates and inactivates certain drugs, especially isoniazid, an anti-tuberculotic drug. These people are called slow acetylators. People with this deficiency have 3 times decreased elimination of the drug compared to normal people. This may increase the risk of toxic effects
- CYP2C9 is an enzyme that around 1% of the population lacks. This causes them to have decreased elimination of drugs like diazepam, making sedation last longer.
- CYP2D6 is an enzyme that 5-10% of the population has too much of, instead of too little. This makes them ultrarapid metabolizers, meaning that they may be non-reactive to drugs that are metabolised by this enzyme, like certain antidepressants and betablockers.
Induction and inhibition of CYP450 enzymes:
Biotransformation enzymes are just like any other enzyme – they may be induced or inhibited. Induction is usually done by increasing gene transcription, while inhibition is usually competitive.
Drugs induce CYP enzymes by binding to receptors that increases gene transcription of the enzyme.
Here is a table of some examples so you understand what I’m talking about:
| CYP family | Drug substrates | Inducer chemicals | Inducer receptor | Inhibitor |
| CYP1 | Caffeine, theophylline, paracetamol | Drug: Omeprazole
Other chemicals: PAHs (found in smoke) |
AHR | Ciprofloxacin, acyclovir |
| CYP2 | Ketamine, warfarin, diazepam, propranolol, codeine | Phenobarbital, rifampin, ethanol | CAR | Clopidogrel, omeprazole, cimetidine |
| CYP3 | Benzodiazepams, HIV antivirals (amprenavir), cyclosporin, testosterone | Rifampin, phenobarbital, spironolactone | PXR | Macrolide antibiotics, ketoconazole, bergamottin (from grapefruit), azole antifungals, ritonavir |
I don’t expect you to know this table, however it makes it easier to come up with examples.
Say you give a patient the protein pump inhibitor omeprazole. Omeprazole will bind to the AHR receptor and increase transcription of enzymes in the CYP1 family. This includes both theophylline and paracetamol, among many others. So, if a patient receives both omeprazole and theophylline the effect of theophylline will be decreased, because the patient has upregulated CYP1 enzymes.
Omeprazole is also an inhibitor of CYP2, so if you gave the same patient warfarin would they be at risk of bleeding out because the levels of warfarin in their blood will be higher than normal.
This table also explains the weird fact of how you shouldn’t take your medication with grapefruit juice. The fruit contains bergamottin, which is an inhibitor of CYP3. Grapefruit juice could therefore increase the effect of drugs that are normally biotransformed by CYP3.
This induction and inhibition may be exploited clinically. Ritonavir and amprenavir are both HIV antivirals, but ritonavir is also an inhibitor of CYP3, which biotransforms amprenavir. By giving both at the same time can we ensure not only increased effectiveness by giving two synergic drugs, but also because ritonavir decreases the elimination of amprenavir.
The same is exploited with ketoconazole, an antifungal, and cyclosporin, an immunosuppressor. Ketoconazole inhibits CYP3 which biotransforms cyclosporin, which means that we can give smaller amounts of cyclosporin to the patient with the same effect. As a bonus ketoconazole prevents fungal infections in the immunocompromised patient.
Hello!
On this statement “Drugs induce CYP enzymes by binding to receptors that increases gene transcription of the drug.” Shouldn’t CYP enzyme inducers increase the gene transcription of more enzymes??
Yes, small brain fart there. fixed now. thanks.