Table of Contents
Page created on January 7, 2019. Last updated on January 7, 2022 at 21:50
Summary
- Certain drugs are bound to proteins in the plasma, mostly albumin
- Drugs that have extensive plasma protein binding have:
- Delayed onset of effect
- Longer duration
- Volume of distribution (Vd) illustrates where in the body the drug is distributed (i.e. only in plasma, in plasma and interstitium, plasma + interstitium + intracellular or inside tissues)
- The higher the Vd value for the drug, the more fluid spaces the drug distributes itself into
- Generally, if Vd is around 0.10 L/kg is the drug only distributed in plasma
- Generally, if Vd is between 0.50 and 1 L/kg is the drug distributed in all fluid compartments
- Generally, if Vd is above 2-3 L/kg is the drug distributed in tissues
- The body burden of the drug is a fancy way to say how much drug is in the body in grams or moles. The body burden can be calculated like this: Body burden = Vd x Cplasma
Drug fate after absorption
After absorption will the drug reach the blood. When in the blood can three things happen to the drug:
- It can bind to plasma proteins like albumin and AGP
- It can bind to components of RBCs
- It can leave the blood to be taken up by tissues
Binding of drugs to plasma proteins
Different drugs bind to plasma proteins with to a different extent and with different strength.
The extent to which the drugs are bound to plasma is expressed as a percentage. For example, paracetamol has a 20% plasma protein binding, meaning that 20% of all paracetamol molecules in the blood are bound to proteins. The remaining 80% is dissolved in the plasma itself.
The strength to which the drugs are bound to proteins is expressed by a constant called KA. For most drugs is the value between 103 – 104 L/mol. It’s higher for certain drugs, like warfarin. This parameter isn’t as important as the protein binding extent.
Drugs that are bound to plasma proteins are not active and can’t leave the blood. This is important to keep in mind.
So which plasma proteins are important here? There are two that are significant, however certain drugs may also bind to other plasma proteins.
Albumin is the most important. It’s abundant in the blood as it has a concentration of 4 g/dL, so it has a high capacity to bind drugs. It mostly binds acidic drugs. It’s a negative acute phase protein, which means that its level is decreased during inflammation.
α1-acid glycoprotein (AGP) is the second most important. It has a much lower concentration, 50 – 150 mg/dl, so it has a lower capacity to bind drugs. Its properties are mostly opposite to those of albumin, meaning that it binds mostly basic drugs and it is a positive acute phase protein, meaning that its level increases during inflammation.
The amount of drug that is protein bound and the amount that is dissolved is always in equilibrium. Let’s say that a drug has 50% plasma protein binding, and let’s say that there are 100 drug molecules in the plasma. 50 drug molecules are bound and 50 are not. If the 50 drug molecules that are not bound will leave the plasma and enter the tissue will 25 of the protein-bound drug molecules loosen from the proteins. This means that there are now 25 drug molecules bound to plasma proteins, 25 that are dissolved in plasma, and 50 molecules in the tissues.
Why is plasma protein binding of drugs important? Several reasons. Drugs that are extensively plasma protein-bound:
- may have a delayed onset of effect
- may have a prolonged effect and delayed elimination
- may displace other drugs that have lower plasma protein-binding
- may displace endogenous compounds that are bound to plasma proteins
- may have increased effect when the level of plasma proteins is low and vice versa
- can’t be efficiently removed by haemodialysis
1 and 2: When drugs bind extensively to plasma proteins will the plasma protein-bound fraction act like a “reservoir” of the drug inside the body. Drug molecules are only slowly released from this reservoir. This is easiest to understand if you consider a drug that has 0% plasma protein binding – this drug would be absorbed, enter the blood and immediately enter the tissues and elicit its effect. This drug would have immediate onset of effect, and its effect would be short-lived. Drugs with extensive plasma-protein binding have the opposite properties, meaning that they will have delayed onset of effect, and their effect will be prolonged.
Consider two different insulin medications. One is engineered so that it has 90% plasma protein binding, and the other has just 10%. The first drug, when administered, will be useful for a sustained insulin release, but it will not be useful in acute situations of hyperglycaemia. For that will the 10% drug be more suitable.
3: Plasma proteins have a limited number of available binding sites that drugs can bind to. Let’s say we give a drug that is 50% plasma bound. We then give another drug that is 90% plasma bound. For the second drug to be 90% plasma bound must it “steal” some plasma protein binding sites from the first drug. This means that as the second drug is administered will the first drug suddenly drop on plasma binding, from 50% maybe down to 20% or something. This means that there is a sudden release of the first drug into the tissues.
This can be dangerous in many cases. Let’s say we first give an antidiabetic medication with 50% plasma protein binding. We then give another drug with 80% plasma protein binding. The antidiabetic will suddenly be released into tissues, potentially causing significant hypoglycaemia.
4: Not only drugs bind to plasma proteins, endogenous compounds like bilirubin and uric acid. These compounds can also be displaced by extensively plasma protein-binding drugs.
Certain antibiotics displace bilirubin, meaning that the level of free bilirubin increases in the blood. This extra bilirubin can cause brain damage in neonates.
This is especially important in renal failure patients. These patients have uraemia, so they have more waste products and other endogenous compounds in the blood than healthy people. Some of these compounds may bind to plasma proteins and displace drugs with scarce plasma protein-binding, which can cause problems.
5: Let’s say you have 100 molecules of albumin and 100 drug molecules in the blood. The drug has 90% plasma protein binding, so 90 of the drug molecules are bound to albumin and 10 are dissolved.
Now, let’s imagine another situation. There is now hypoalbuminaemia, so there are only 50 molecules of albumin available but still 100 drug molecules. The drug still has 90% plasma protein binding. 90% of 100 drug molecules is 90 drug molecules, so these 90 drug molecules want to bind to albumin. However, there are only 50 molecules of albumin now. This means that only 50 drug molecules can bind to albumin, while the remaining 45 must remain dissolved in the plasma together with the 5 molecules that didn’t want to bind to albumin. Instead of a 90% of the drug molecules being bound to albumin are only 50% of them bound.
This shows that when plasma protein level is low will extensively plasma protein-binding drugs have a larger effect. The opposite is true when the level is high – the effect will be lower. This is especially important for drugs that bind to AGP, because AGP levels are increased during inflammation. This means that drugs that bind to AGP in the plasma have decreased effect during inflammation.
6: Haemodialysis removes molecules from the blood that shouldn’t be there. It’s often used in intoxications and kidney failure. However, if a large fraction of the drug is bound to plasma proteins will it be harder to remove by dialysis, because plasma proteins (and drugs that are bound to them) are not removed by dialysis. Only smaller molecules are.
Binding of drugs to RBCs
A few drugs bind to carbonic anhydrase or a protein called immunophilin in RBCs. It’s not so important.
Distribution of drugs to tissues
We must introduce a new term – volume of distribution. The symbol for it is Vd.
The Vd of a drug represents the drug’s tendency to distribute into different volume spaces in the body. It’s expressed in L/kg, i.e. how many litres of body fluid per kg body weight you would need to distribute the drug.
Some sources, like BRS and Wikipedia, express Vd in just L instead of L/kg. That is because those sources calculate the Vd for an average person weighing 70 kg. So to convert values in L/kg to L you should multiply by 70. In my opinion it makes more sense to give the value in litres, but anyway. This is definitely not important for the exam.
The concept of Vd can be hard to grasp, but it’s useful because it can be used to determine how the drug will distribute in the tissues of the body. If a drug has a low Vd does that indicate that the drug mainly stays in the plasma. A very high Vd (several L/kg) indicates that the drug accumulates in tissues.
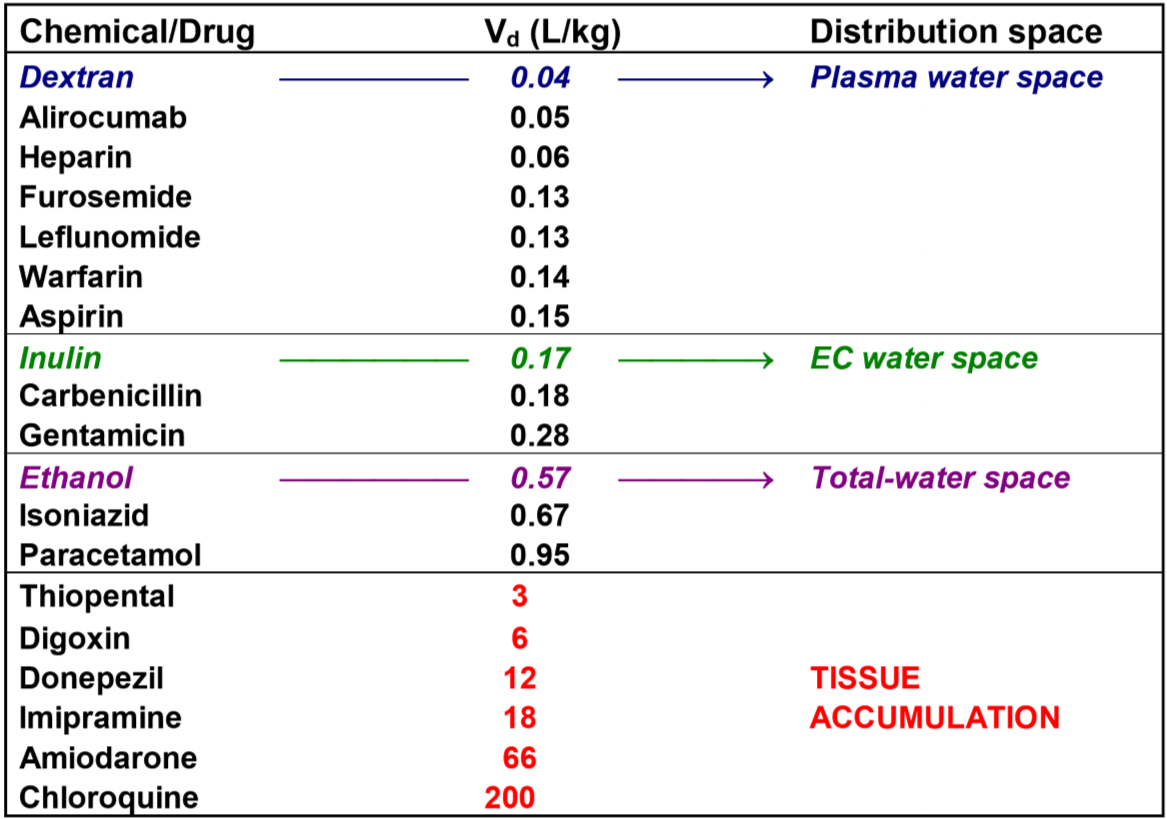
An average person of 72 kg has approx. 3 litres of plasma fluid. 3L/72kg = 0.04 L/kg, which means that 4% of the total body weight is plasma fluid. The Vd of a drug called dextran is 0.04 L/kg. Dextran is a very large molecule. It’s so large that it never leaves the plasma, meaning that dextran disperses into the whole plasma fluid space and not at all into other fluid spaces. It’s no coincidence that the Vd of dextran is the same as the plasma fluid volume – it’s because dextran disperses into the plasma fluid and nowhere else.
The water content in an average person’s body is 60% of the total body weight, so a 72 kg person has around 43 L of total water. 43L/72kg = 0.60 L/kg. The Vd of ethanol is 0.57 L/kg. The reason the Vd of ethanol is so similar to the total water content of the body is because ethanol diffuses into all water spaces of the body – intracellular and extracellular.
Generally can we say that:
- If a drug has Vd between 0.04 and 0.15 L/kg does it distribute in the plasma fluid space
- If a drug has Vd between 0.17 and 0.27 L/kg does it distribute in the whole extracellular fluid space
- If a drug has Vd between 0.57 and 0.95 L/kg does it distribute in the whole water space, intracellular and extracellular
- If a drug has Vd above 3 L/kg does it accumulate in tissues
Here is a chart that maybe makes that clearer. It’s not important to know by hand.
Then, what determines a drugs Vd? Many factors:
- The size of the molecule. Very large molecules stay in the plasma. Large molecules stay in the plasma and extracellular space. The larger the molecule, the lower the Vd.
- The degree of plasma protein binding. The higher the degree of binding, the more drug stays in the plasma, giving a lower Vd
- The solubility of the drug. Hydrophilic drugs mainly distribute into the extracellular space. Lipophilic drugs distribute in the tissues as well. Very lipophilic drugs may accumulate in adipose tissue.
So, the Vd determines which fluid compartment a drug will accumulate in. Then, what determines which organs it will accumulate in? Three things: The blood flow the organ receives, the “tissue affinity” of the drug and the capillary porosity of the organs.
Drugs accumulate initially in organs with the highest blood flow. In descending order, the important organs that receive the most blood flow are: Lungs, kidneys, heart, brain and liver. However, blood flow only determines where drugs initially accumulate.
Drug will later redistribute into organs that is characteristic for the drug. For example, thiopental redistributes into the muscle and adipose tissue, while chloroquine redistributes into the liver.
Different organs have different capillary beds with different pores between the endothelial cells. Certain organs even have fenestrae, which are not paracellular pores but pores that are transcellular. Here is a table:
| Capillary bed | Pore size (nm) | Pore permeable for |
| Hepatic sinusoids
Renal peritubular capillaries Spleen, bone marrow capillaries |
50 – 200 (fenestrae) | Proteins, protein-bound drugs, unbound drugs |
| Renal glomerulus
Muscle |
5 – 8 | Unbound drugs |
| Brain | 0 | None (blood brain barrier) |
Because the spleen, bone marrow, liver and kidney have the largest pores for drugs to pass through is it understandable that these organs are common places where drugs accumulate.
Distribution of drugs in the liver
Accumulation in the liver is common because:
- Drugs given orally always pass through the liver (first pass metabolism)
- The hepatic sinusoids have large pores
- The sinusoidal membrane of liver cells is designed to take up drugs, so they have microvilli and many drug transporters
Two types of drugs especially accumulate in the liver:
- Cationic amphiphilic drugs (because they become trapped inside lysosomes)
- Lipid soluble vitamins (are stored inside the Ito cells / stellate cells)
Distribution of drugs in the lungs
The lung is exposed to many drugs, at least transiently, because:
- The lung has the largest blood flow of all organs
- Drugs given parenterally (not by mouth) pass through the lung first
Later may drugs redistribute to their favourite organ, as explained above.
Cationic amphiphilic drugs may accumulate in the lungs as well.
Distribution of drugs in the adipose tissue
Drugs that are highly lipophilic accumulate and are stored in fat.
Distribution of drugs in the bone
The main component of bone is calcium apatite, a salt comprised of Ca2+, PO43- and OH–. Certain drugs may be deposited into this salt. The best example of this is tetracycline, which gives bone and tooth problems in young children because of this.
Drugs may also be incorporated in calcium apatite instead of the calcium, phosphate or hydroxide ions that are supposed to be there.
Distribution of drugs in the skin
Certain drugs are keratophilic, meaning that they bind to keratin inside keratinocytes, causing them to accumulate in the skin.
Distribution of drugs in the thyroid gland
Thyroid follicular cells accumulate I– for the synthesis of T3 and T4. This can be exploited clinically. For example can hyperthyroidism be treated by giving the patient a radioactive isotope of I–. The follicular cells will happily take in the radioactive iodide, which will then kill parts of the thyroid with radiation.
Weaker radioactive iodide isotopes are used for thyroid diagnostics. Certain other antithyroid drugs also accumulate in the thyroid.
Distribution of drugs in the brain
The brain protects itself from harmful substances with the blood-brain barrier. This barrier has multiple components:
- Tight junctions between capillary endothelial cells, so that no molecules can enter through the paracellular route
- No fenestrae in the endothelial cells
- The basement membrane, astrocytes and pericytes fortify the barrier by acting as extra protecting layers
- The endothelial cells have drug exporter proteins like Pgp, BCRP and MRP that actively pump out any drug molecules that managed to sneak through
So, what type of drugs can enter the brain? Such drugs must have either of these characteristics:
- Be lipid soluble or amphiphilic
- Be slightly lipid soluble, small and with scarce plasma protein-binding
- Have a protein transporter that transports the drug across the blood-brain barrier
- Lactate, pyruvate, ketone bodies are transported by MCT1
- LAT1 transports amino acids and amino acid-like drugs, like methyldopa and L-DOPA
That means that this type of drugs generally can’t enter the brain:
- Drugs that are hydrophilic (unless carried by transporters)
- Drugs that are proteins
- Drugs that are highly plasma protein-bound
- Drugs that are substrates for the drug exporter proteins Pgp, BCRP and MRP
Certain parts of the brain are not protected by the blood brain barrier. These organs are called the circumventricular organs are includes most importantly area postrema, which is involved in vomiting and nausea. This means that drugs can cause nausea and vomiting even if they don’t cross the blood brain barrier. Antiemetic drugs take advantage of this.
Sometimes we need to promote entry of a certain drug to the brain. We can do this by giving hypertonic mannitol into the carotid artery simultaneously with the drug. This causes the endothelial cells to contract, which widens the tight junctions between them so that drugs may pass through. This is especially used for anticancer drugs used for brain tumors.
Drug distribution across the placenta and into the foetus
The placenta acts like a barrier between maternal and foetal blood. There are three ways drugs can cross this barrier:
- Diffusion
- Carrier-mediated transport
- Receptor-mediated endocytosis
Because drugs can diffuse across the barrier can we understand that the barrier does not protect against lipophilic drugs at all. The barrier is only effective against hydrophilic drugs, highly protein-bound drugs and drugs that are substrates for the drug exporters Pgp, BCRP and MRP.
REffereing to what you said in point 5: if theres only 50 albumin and the drug has a 90% plasma protein binding, then 45 of them will be bound to albumin. The drug does not know that the patient has hypoalbuminemia. So it is 45. Not 40.
fixed now
Can you give a reference for the fact that hepatic sinusoids, bone marrow, etc. allow protein bound drugs to penetrate into bone marrow?
It was taken directly from our lecture on the topic, however I can’t seem to find any good sources for it. Skimming this study even makes it look like exogenous macromolecules enter the bone marrow by phago-endocytosis, and that transcellular transport of bone marrow sinusoids is used for cells.
The lecture has the following paragraph related to this:
“Fenestrae are transcellular pores in the endothelial cells, i.e. the cell bodies are perforated. These
large pores permit the passage of even protein-bound drugs to reach the hepatocytes (from the
hepatic sinusoids) or the proximal tubular cells (from the renal peritubular capillaries), and permit
antibodies against B cells (e.g. rituximab) and myeloid cells (e.g. gemtuzumab) to reach their target
cells in the spleen and the bone marrow. In contrast, the pores in the glomerulus and muscle
capillaries are paracellular.”
I tried to look for information on how gemtuzumab enters the bone marrow, but I couldn’t find anything. Maybe you’ll have more luck.
Sorry, I’m so confused. how is it possible that plasma-bound drugs are always in equilibrium with their free unbound counterparts, but at the same time we say that drugs can have higher or lower plasma protein binding. If we have 90% plasma protien binding of a drug, that only leaves the 10% fraction of free drug molecules. This doesn’t sound like equilibrium
It means that, no matter how much drug or how much protein we have, 90% of the drug molecules in the plasma will be bound to proteins and the rest will be free.
If we have 100 drug molecules in the plasma 90 drug molecules are bound to proteins and 10 are free. If the 10 free drug molecules are eliminated somehow, we will have 90 protein-bound drug molecules and 0 free ones. This pushes the equilibrium towards the free fraction, which causes 9 protein-bound drug molecules to dissociate from proteins. The situation will then be that there are 9 free drug molecules and 81 plasma protein bound drug molecules. Once again 90% of the drug molecules in the plasma are bound to proteins while the remaining 10% are free.
“Proteins that have extensive plasma protein binding have” –> assume its supposed to be drugs not proteins in the beginning of the sentence? 🙂
fixed