Table of Contents
Page created on February 16, 2019. Last updated on January 23, 2023 at 18:54
Physiology recap
General information
Thrombosis is the pathological formation of a “haemostatic” plug within the vasculature in the absence of bleeding. We wish to prevent thrombosis as much as possible, to prevent embolism and ischaemia. A small refresher of haemostasis physiology may be helpful.
Haemostasis is the process that causes bleeding to stop when blood vessels are damaged. The process involves three steps:
- Vasoconstriction
- Adhesion and activation of platelets to form a platelet plug
- Fibrin formation
In addition to this, the fibrin be will removed by fibrinolysis after the bleeding has stopped.
Haemostatic drugs affect haemostasis by interfering with step 2, 3 or by decreasing the rate of fibrinolysis. This topic will only cover drugs that interfere with platelet function and fibrin formation.
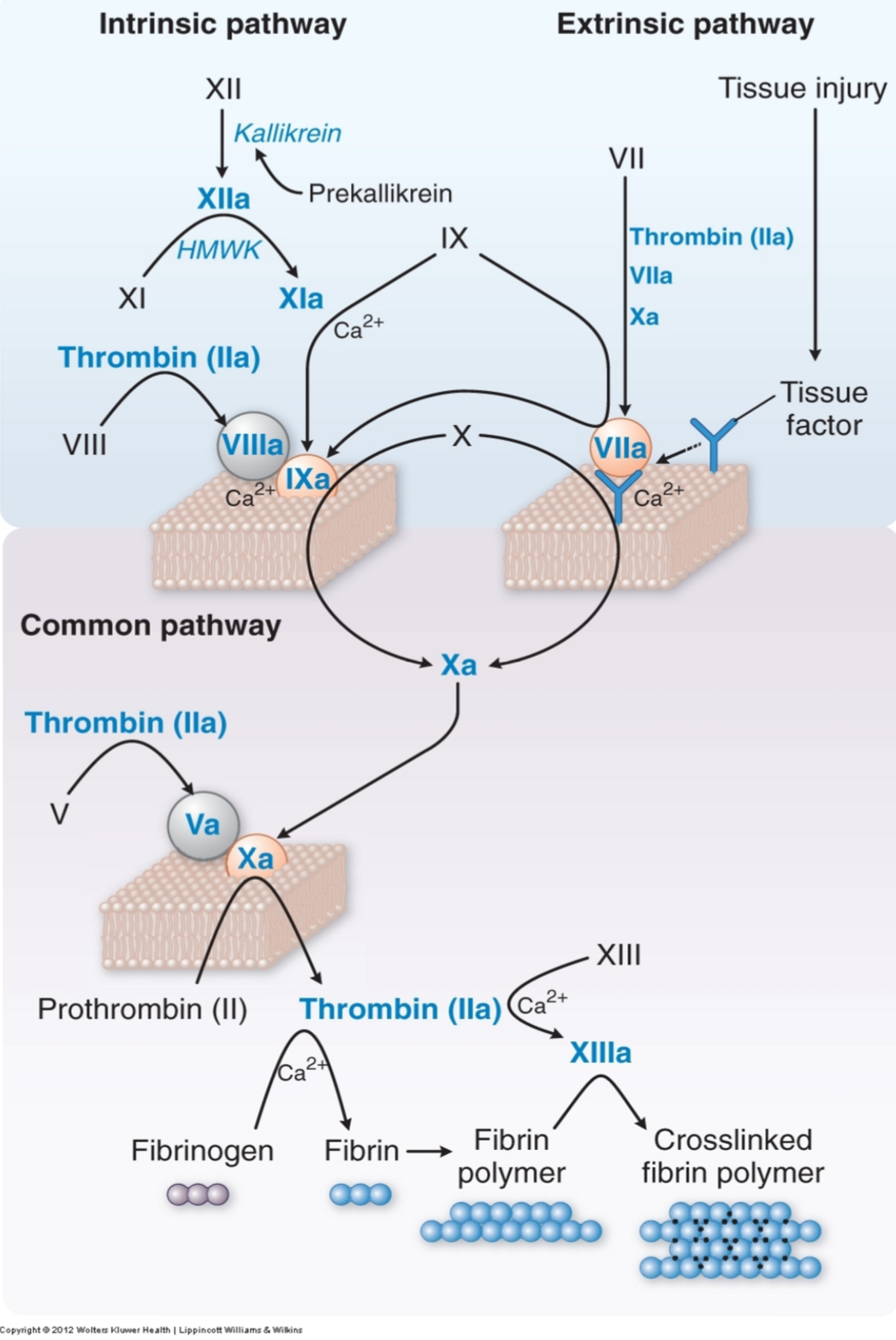
There are two pathways that lead to the formation of fibrin: the intrinsic pathway and the extrinsic pathway. Both end in the same pathway, the common pathway.
The intrinsic pathway is so named because all the components of it can be found in the blood. A better and more modern name of the pathway is the “contact activation pathway”, because it is activated when the blood comes into contact with a negative surface like glass or another foreign surface. Factor XII binds to the foreign surface and becomes activated to XIIa, which then converts XI into XIa which converts IX into IXa which converts X into Xa.
The extrinsic pathway is so named because it involves a factor that isn’t present in the blood, the tissue factor. Tissue factor is a protein that is found in subendothelial tissue. When there is a vessel damage will the subendothelial tissue and therefore tissue factor be exposed to the blood. Tissue factor activates factor VII to VIIa, which converts X into Xa.
Both pathways end when the common pathway starts, when Xa is formed. Xa converts prothrombin into thrombin, which again converts fibrinogen into fibrin. Fibrin then cross-links with the help of factor XIIIa to form a fibrin mesh that covers the vascular defect.
Measurement of clotting status
There are multiple laboratory tests that test the functions of these pathways. Prothrombin time and international normalized ratio (INR) both test the function of the extrinsic pathway while the partial thromboplastin time (PTT) tests the function of the intrinsic pathway.
Important factors
Vitamin K
Vitamin K is a fat-soluble vitamin that is essential for the production of clotting factors II, VII, IX, X and protein C and protein S in the liver. After the translation of these clotting factors they are post-translationally modified by γ-carboxylation. This enables them to bind Ca2+, which is essential for their function. In the absence of vitamin K these clotting factors will still be synthesized, but they won’t function as they can’t bind Ca2+.
Vitamin K isn’t strictly a cofactor for γ-carboxylation, as it is converted to an unusable form in the process. However it can be recycled so it can be reused. Recycling is done by the enzyme vitamin K epoxide reductase. When this enzyme works properly, vitamin K will be continuously recycled and reused.
Antithrombin III
Antithrombin III is an endogenous molecule that inhibits factors II, IX, X, XI and XII and therefore the formation of fibrin.
Anticoagulant drugs
Introduction
Anticoagulant drugs are widely used in medicine, both in internal medicine and surgery. Many patients are on anticoagulants. Anticoagulant drugs are never combined.
Types
- Vitamin K antagonists (VKA)
- Unfractionated heparin (UFH)
- Low molecular weight heparin (LMWH)
- Direct oral anticoagulants (DOAC)
Indications
Anticoagulants are mostly used to prevent venous thrombosis, and in higher doses, treat them. These are the most important indications:
- Prevention of stroke in case of atrial fibrillation
- Mechanical prosthetic heart valve
- Acute myocardial infarction
- Nephrotic syndrome
- Prophylaxis for venous thromboembolism (DVT, PE)
- Perioperative prophylaxis
- Prophylaxis in case of immobilisation
Historically, vitamin K antagonists were generally used for longer-term anticoagulation, while unfractionated heparin was generally used for shorter-term anticoagulation. Nowadays, DOACs have mostly replaced the use of vitamin K antagonists, and LMWHs have mostly replaced the use of unfractionated heparin.
Most of the mortality and morbidity of atrial fibrillation comes from the fact that the inefficient atrial contractions predispose to the formation of thrombi, which can embolise and cause stroke. Many people with atrial fibrillation require indefinite anticoagulation to prevent this.
Adverse effect
The major adverse effect of anticoagulation is an increased risk of bleeding, especially intracranial bleeding in case of falls, and spontaneous gastrointestinal bleeding from ulcers or other lesions. These bleedings can be fatal.
Vitamin K antagonists
Compounds
- Warfarin (Marevan®, Coumadin®)
- Dicumarol
Most vitamin K antagonists (VKAs) are coumarins, so the terms are often used interchangeably. However, there exist some VKAs which are not coumarins. The most commonly used VKA by far is warfarin. These drugs are administered orally.
Following the introduction and establishment of DOACs (see below), VKAs are much less commonly used, as DOACs are safer and non-inferior to VKAs in most cases. There are now only a few cases where VKAs are preferred compared to DOACs:
- Mechanical prosthetic heart valve
- Moderate/severe mitral stenosis
Mechanism of action
VKAs inhibit vitamin K epoxide reductase, thereby decreasing the amount of “usable” vitamin K. This creates a condition similar to vitamin K deficiency, causing clotting factors II, VII, IX and X to be dysfunctional and therefore inactive. This creates a functional depletion of these clotting factors.
Because VKAs act on the synthesis of clotting factors they have a slow onset of action. Immediately after the first administration there are still functioning clotting factors present in the blood – the anticoagulant effect comes only after these functioning clotting factors have been eliminated, which takes around 4 days.
Pharmacokinetics
Warfarin has good oral absorption, strong plasma protein binding (97%) and is inactivated by CYP450 in the liver. Its duration of action is 4-5 days and the half-life is 40 hours.
Monitoring
The problem with coumarins is that people respond differently to the same dose. This is because polymorphisms in the gene for vitamin K epoxide reductase change the affinity of the VKAs to the enzyme and differences in vitamin K content of diet.
It’s therefore important to start with a low dose and continuously test the INR of the patient to make sure that they’re not receiving too much (and therefore bleed too easily) or too little (and therefore have suboptimal anticoagulant effect). The dose should always be adjusted so that the INR of the patient is between 2 and 3 (between 2,5 – 3,5 in case of mechanical heart valves). INR should be measured often, initially daily but later more and more rarely (but still regularly).
Contraindications
VKas are teratogenic and are therefore contraindicated in pregnancy. They’re also contraindicated in severe liver and kidney failure.
Interactions
Certain drugs decrease the effect of coumarins. Vitamin K is the obvious one – in fact vitamin K is the antidote to warfarin poisoning. Drugs that induce CYP450 enzymes like rifampicin and barbiturates increase the elimination of coumarins. Colestyramine decreases the absorption of them.
Because coumarins have strong plasma protein binding they can be displaced by other drugs who also bind strongly to plasma proteins.
Complications
Warfarin inhibits the synthesis of protein C and protein S as well, and these factors are depleted more quickly than the clotting factors. This causes an initial hypercoagulable state which can cause tissue infarction and necrosis. Warfarin-induced skin necrosis is a severe complication which can occur with warfarin treatment.
To prevent warfarin-induced necrosis we can use heparin to prevent the initial hypercoagulable state.
Reversal
The anticoagulant effect of VKA antagonists can be rapidly reversed by replacement, by administering prothrombin complex concentrate or fresh frozen plasma. Vitamin K administration also antagonises the effect but this occurs much more slowly.
Direct oral anticoagulants
Compounds
- Direct thrombin inhibitors
- Dabigatran etexilate (Pradaxa®)
- Argatroban
- (NB! Not taken orally, so not technically a DOAC)
- Direct factor X inhibitors
- Apixaban (Eliquis®)
- Rivaroxaban (Xarelto®)
- Edoxaban
- Antidotes
- Idarucizumab (antidote to dabigatran)
- Andexanet (antidote to apixaban and rivaroxaban)
These drugs are called direct oral anticoagulants (DOACs) as they act directly on one of the clotting factors and because they’re taken orally (except argatroban). They were previously abbreviated NOACs (N for “novel”), although they were introduced more than 10+ years ago, so this name is no longer fitting.
DOACs are mostly used as safer and non-inferior alternatives to warfarin.
Mechanism of action
These drugs bind to and inhibit certain clotting factors directly. Dabigatran and argatroban bind to and inhibit thrombin (factor IIa). Rivaroxaban, apixaban, edoxaban are factor Xa inhibitors.
Idarucizumab is a monoclonal antibody which binds to and inactivates dabigatran. Andexanet is a modified recombinant factor Xa. Apixaban and rivaroxaban bind to andexanet with the same affinity as factor Xa, which frees up endogenous factor Xa.
Pharmacokinetics
DOACs are excreted renally, and so require dose adjustment in mild forms of kidney failure and possibly being contraindicated in severe forms. Dabigatran is renally excreted to a much larger degree than the factor Xa inhibitors.
However, they’re also metabolised by the liver and so are also contraindicated in severe liver failure.
Contraindications
- Pregnancy
- Mechanical prosthetic heart valve
- Severe kidney failure
- Severe liver failure
- Extremes of body weight (very high or very low)
- Requires dose adjustment in some DOACs and is contraindicated for others
Reversal
Idarucizumab and andexanet can be used to reverse their effect, but are not readily available and are quite expensive.
Advantages of DOACs vs VKAs:
- Don’t require regular monitoring
- Can be taken orally
- Lower bleeding risk
Disadvantages of DOACs vs VKAs:
- Reversing their effect requires antidotes which are very expensive and not readily available
- More expensive
Unfractionated heparin
Compounds
- Unfractionated heparin (UFH)
- Protamine sulphate (antidote)
The term “unfractionated” is used to separate it from LMWH (see below), although UFH is frequently called simply “heparin”.
Heparin isn’t actually a single molecule but a family of large and sulphated glycosaminoglycans that all act on antithrombin III. Heparin is actually present endogenously in the body inside the granules of mast cells. To acquire heparin the pharmaceutical industry extracts them from beef lung or pig intestine. However, because heparin isn’t a single molecule can the biological activity of it differ depending on where it is extracted from. Because of this the dose of heparin is not given in units of mass but rather in units of activity.
The molecular weight of heparin is between 5 and 35 kDa, depending on where it was extracted from.
Indications
Heparin is preferred over warfarin in cases where it is necessary that the anticoagulant effect begins immediately. It’s used to prevent/treat deep vein thrombosis, pulmonary embolism, and acute coronary syndromes.
Heparin is administered IV or s.c.
Mechanism of action
Heparin increases the effect of antithrombin III and therefore has strong anticoagulant activity.
Pharmacokinetics
It acts immediately when given IV but has a 60-minute delay when given subcutaneously. Because of its large molecular size heparin would not be absorbed through the GI tract.
Heparin is eliminated by the liver and by phagocytosis by macrophages. It’s safe to use in pregnancy.
Monitoring
Like for warfarin must patients receiving heparins measure its effect frequently to ensure that they’re not overtreated or undertreated. Activated partial thromboplastin time (aPTT) should be measured daily and should be 1.5 – 2.5 times that of a control subject.
Side effects
Heparin may paradoxically cause thrombosis. It’s an uncommon but serious side effect called heparin-induced thrombocytopaenia (HIT). It occurs if IgG and IgM antibodies are produced against heparin and platelet factor 4. The immune complexes that form activate platelets and cause thrombosis. Opsonized platelets are phagocytosed, causing thrombocytopaenia.
HIT is treated by taking the patient off heparins and giving another anticoagulant instead, most commonly either danaparoid or argatroban. Platelet transfusion may be necessary. Warfarin can not be used due to it’s slow onset of action.
Antidote
The antidote of heparin is a drug called protamine sulphate, which can be used if there is excessive bleeding. It binds to and inactivates heparin.
Low molecular weight heparins
Compounds
- Enoxaparin (Clexane/Klexane®)
- Dalteparin (Fragmin®)
- Fondaparinux
The low molecular weight heparins (LMWH) are fractionated heparins as opposed to unfractionated heparin. These drugs are just fragments of the unfractionated heparin. They’re more predictable and have longer half-life than unfractionated heparin and are therefore preferred in most cases.
The LMWHs have a molecular weight of 3 – 4 kDa.
Mechanism of action
LMWHs don’t inhibit all the coagulation factors that unfractionated heparin does; they only inhibit Xa. However that’s more than enough to ensure that LMWHs are at least as safe and effective as unfractionated heparin and are more convenient to use.
Advantages of LMWH vs unfractionated heparin
- No monitoring of APTT or other parameters is necessary as they’re more predictable
- Patients can be taught to inject themselves subcutaneously at home
- Lower risk of bleeding
- Lower risk of heparin-induced thrombocytopaenia
Disadvantages of LMWH vs unfractionated heparin
- LMWH are excreted renally and therefore cannot be used in severe renal failure
Antiplatelet drugs
Introduction
As with anticoagulants, antiplatelets are also widely used in medicine (mostly cardiology and vascular surgery). Many patients are on antiplatelets.
When there is a vascular damage receptors receptors on the surface of platelets will bind to the subendothelial collagen with the help of von Willebrand factor. This causes the platelets to change shape and release their granules, which contain thromboxane A2, ADP and serotonin.
The molecules that were released from the granules bind to receptors on other platelets and activate them too. ADP will bind to P2Y12 receptor (and the P2Y1 receptor). During platelet aggregation, the glycoprotein IIb/IIIa receptor on the platelet surface binds to fibrinogen. This aggregates the platelets by sticking them together.
By interfering with the platelet’s function in haemostasis one can also inhibit thrombosis. Different drugs act on different aspects of platelet activation and aggregation.
Types
- Acetylsalicylic acid
- P2Y12 receptor antagonists
- Dipyridamole
- Glycoprotein IIb/IIIa inhibitors
Indications
Antiplatelets are mostly used to prevent arterial thrombosis, and in higher doses, treat them:
- Acute myocardial infarction (treatment and prophylaxis)
- Ischaemic stroke (prophylaxis)
- After percutaneous coronary intervention (to prevent thrombosis of the stent)
- Peripheral artery disease
As you can see, antiplatelets are mostly used in case of arterial thrombosis due to atherosclerotic cardiovascular disease.
Acetylsalicylic acid
- Acetylsalicylic acid (ASA, Albyl-E®)
ASA is technically an NSAID, although it’s more frequently used as an antiplatelet rather than for its anti-inflammatory effects. It’s a very frequently used antiplatelet.
ASA is given in much lower dose for use as an antiplatelet drug than when used as an anti-inflammatory. Antiplatelet dose is 50 – 150 mg/day and in an extended-release formulation (Albyl-E®) while anti-inflammatory dose is 500 mg/day (Aspirin®). ASA has no anti-inflammatory effect in these low antiplatelet doses (although it does have an antiplatelet effect in the anti-inflammatory dose).
For more information on ASA in general and as an anti-inflammatory drug, see topic 36.
P2Y12 receptor antagonists
Compounds
- Clopidogrel (Plavix®)
- Prasugrel (Efient®)
- Ticagrelor
P2Y12 receptor antagonists are often used in combination with aspirin, which is called dual antiplatelet therapy (DAPT). However, they may also be used alone.
Mechanism of action
These drugs inhibit the P2Y12 receptor, an ADP receptor which is found on platelets. This receptor binds ADP which is released from other activated platelets. By blocking the P2Y12 receptor these drugs prevent platelet aggregation.
Pharmacokinetics
Clopidogrel and prasugrel are irreversible inhibitors, while ticagrelor is a reversible inhibitor. Despite this, they appear to have similar half-lives.
Glycoprotein IIb/IIIa inhibitors
Compounds
- Abciximab
- Eptifibatide
- Tirofiban
These antiplatelets are mostly used for short-term treatment before and during percutaneous coronary intervention.
Mechanism of action
These drugs bind to and inhibit the receptors glycoprotein IIB/IIIA, which prevents platelets from binding to each other and therefore preventing aggregation.
Dipyridamole
Dipyridamole is often combined with aspirin.
Mechanism of action
Dipyridamole is phosphodiesterase inhibitor. It increases the amount of cAMP in the platelets, which inhibits platelet aggregation.
When to use what?
Both antiplatelet drugs and anticoagulants prevent blood from clotting, so when do we use which type?
The short version of it is that anticoagulants is to prevent thrombosis on the venous side (including the heart) and antiplatelets are used to prevent thrombosis on the arterial side.
Venous thrombi and thrombi forming inside the heart due to atrial fibrillation are mostly due to slow blood flow, or stasis. Stasis activates the coagulation cascade prematurely because the pro-coagulant factors in the blood aren’t exposed to as many anti-coagulant factors on the endothelial surface. In these cases the best treatment is to inhibit the coagulation cascade.
Arterial thrombi form mostly on foreign surfaces like atherosclerotic plaques and stents. This is because the platelets are mostly involved in this process. Antiplatelets decrease the incidents of strokes and coronary episodes like AMI because they prevent platelets from aggregating on the (ruptured) atherosclerotic plaque.
Dear Nik,
incase of platelets aggregation, the correct mechanism is GP Ia/IIb binds to collagen and GP Ib bind to vWF of the damaged sub endothelial tissue, and factor GP IIb/IIIa are used to fibrin linkage between activated platelets and not for binding to the collagen as it is stated on that part of the note.
Thank you for your great notes overall, keep up the good work ✌🏻🙌🏻
Thank you for the correction. Glad you like the notes!
Hi Nik, any reason for that you didnt include direct thrombin inhibitors, e.g. hirudin or bivalirudin ?
Notes are based mostly on the lectures and seminars. When I had pharma 2 the seminar included only one sentence about direct thrombin inhibitors:
“Management of patients with thromboembolic disease who develop HIT is therefore usually with either danaparoid or with a direct thrombin inhibitor (argatroban).”
Hirudin and bivalirudin are not mentioned at all, but argatroban is, so I’ve added that to the topic now.
Hemostatic drugs inhibit fibrinolysis and promote coagulation 🙂
I assume you’re referring to the mistake in the introduction? I’ve fixed that now.
Nice!! Keep up the good work:D
Thank you, you too!
Is there a reason why you haven’t mentioned Heparinoids in this topic?
Added a small note than one heparinoid is used in HIT.
Hi I think there is antidote for Dabigatran, which is Idarucizumab.
Interesting. Added to the topic.