Table of Contents
Page created on September 27, 2018. Last updated on January 7, 2022 at 21:47
Summary:
- Competitive antagonism is when the antagonist binds to the same binding site on the receptor as the agonist or endogenous ligand. This binding can be reversible or irreversible.
- Allosteric antagonism is when the antagonist binds to an allosteric binding site on the receptor, decreasing the effect of the agonist
- Antagonism by inhibiton of signal transduction is when the antagonist inhibits the signal transduction pathway of the agonist
- Functional antagonism is when the antagonist exert effects on the cell which are opposite effects of the agonist
- Pharmacokinetic antagonism is when the antagonist decreases the absorption or increases the elimination of the agonist
- Chemical antagonism is when the antagonist chemically binds to the agonist and inactivates it
- The margin of safety or therapeutic index is the ratio of TD50/ED50. It tells us how safe the drug is at the therapeutic dose
Combined effects of drugs
When we give two drugs (or one drug and one endogenous agonist) together the result can be one of three types. Let’s imagine that drug A has an “effect” of 10 and drug B has an “effect” of 3. When we give both drug A and B to a patient the total “effect” can be up to 13. This is called addition, that the effects of drugs are added together, but not higher than the sum of the effects of the individual drugs. The second case is potentiation, when the combined effect of drugs A and B is more than 13, meaning that the effect of the two drugs together is higher than the sum of the two drugs. The last case is antagonism, when the effect of both drugs together is lower than either of the drugs’ effect. In our case the total effect of drug A and B would be 9 or lower, meaning that drug B effectively inhibits drug A.
The clinical use of antagonists is to reduce the effect of the endogenous agonist. For example, atropine is given to reduce the normal effect of acetylcholine on muscarinic receptors in the body. Usually (and logically), the more antagonist we give to the patient, the lower the effect of the endogenous agonist.
There are multiple types of antagonists. Let’s look at them.
Reversible competitive antagonists
This is the most common type of antagonism. It’s “competitive” because the antagonist binds to the same site on the receptor as the agonist, and “reversible” because the antagonist binding is reversible. When we give both an agonist and this type of antagonist to a patient, the two drugs will compete for the agonist-site on the receptor. The concentrations of the agonist and antagonists, and their affinities (Kd value) determine which one of the two will occupy the most receptors.
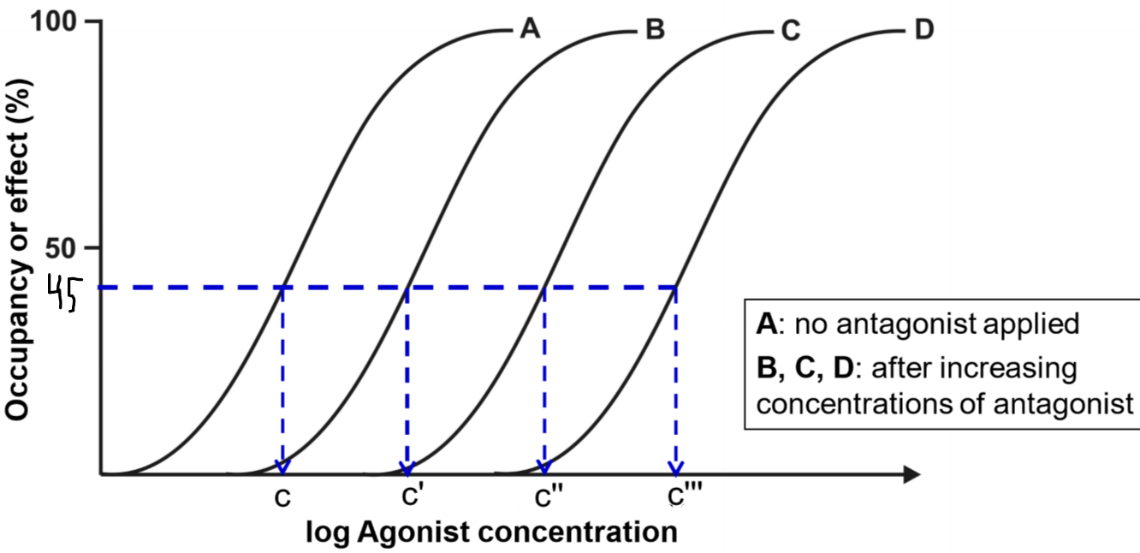
This figure shows four drug-response curves. They are all from the same drug. Curve A shows the drug-response relationship of the agonist when there is no antagonist present. We then apply a low concentration of the antagonism first and then create another drug-response curve, we get curve B. This curve has the exact same shape but is shifted to the right. Then we increase the concentration of the antagonist and do the same again and again, giving curves C and D.
From the figure, we can see that when there was no antagonist present (curve A), the concentration needed to achieve 45% effect was concentration c. For curve B, when there was a low concentration of antagonist present the concentration needed for the same effect was c’, which is higher than c. This means that the higher the concentration of the antagonist, the higher concentration of agonist is needed to achieve the same effect as without the antagonist.
The degree of antagonism shows how much the drug-response curve shifts to the right when the antagonist is applied. Degree of antagonism is denoted by the constant r, called the concentration ratio. This becomes important soon.
Reversible competitive antagonism is surmountable. It means that despite what we saw in the previous paragraph, the agonist may still reach the full effect (100%) after the antagonist is applied, as long as a large enough dose of agonist is applied.
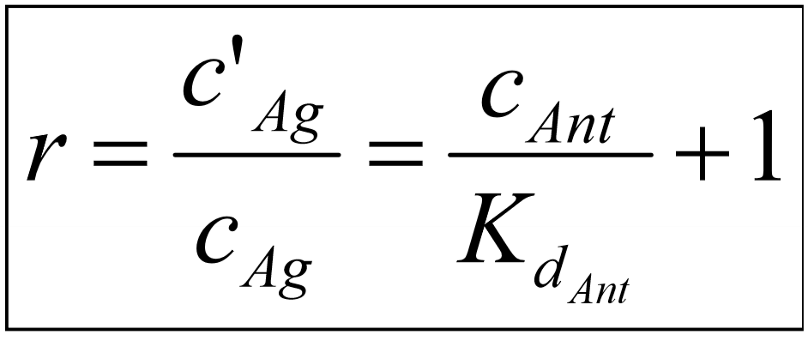
This equation shows that the degree of antagonism (the concentration ratio, r) depends on the concentration of the antagonist and its affinity to the receptor. We invent a new parameter and call it pA2. pA2 is calculated like this:
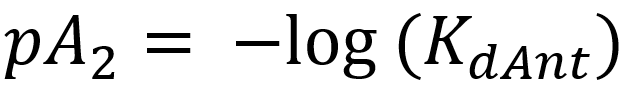
For antagonists, we use the pA2 value to show its affinity.
It’s also important to note here that partial agonists can also act as antagonists. The endogenous ligand is often a full agonist. If we add an exogenous partial agonist to the mix will the partial agonist bind to some of the receptors instead of the endogenous full agonist. This results in an overall reduction in “agonist” activity.
Irreversible competitive antagonists
This type of antagonist is also competitive, but the drug-receptor binding is irreversible, meaning that when a drug first has bound to the receptor it will never un-bind. This binding is often covalent. Irreversible competitive antagonists will exert their effects until the body has had time to produce new receptors, which often takes days.
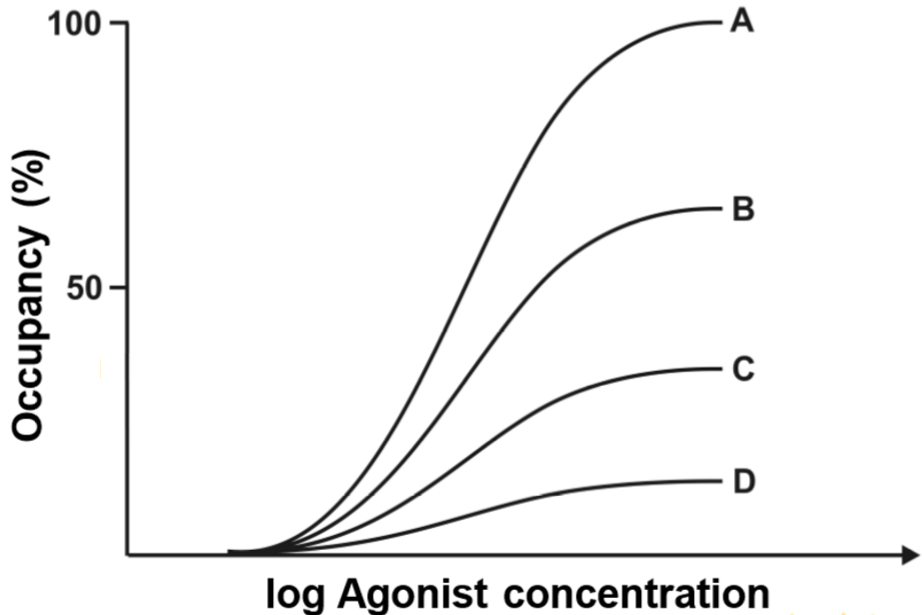
This figure shows four drug-occupancy curves. Curve A was calculated when there was no irreversible antagonist present. Curve B, C and D were calculated when there were increased concentrations of irreversible antagonist. The graph shows that, quite logically, when increasing amounts of irreversible antagonist is applied, the occupancy of agonist decreases. This is because the antagonist occupies a fraction of the available receptors, so that the number of receptors that are available for the agonist to bind to decreases.
When curve B was drawn, the maximum occupancy of the agonist was approximately 60%. This means that the antagonist had occupied 40% of the total number of receptors. When curve D was drawn the agonist occupancy was around 15%, so the antagonist had occupied 85%.
The graph above showed how the occupancy of the agonist changed when an antagonism was applied. Now let’s look at how the effect of the agonist is changed in presence of an antagonist.
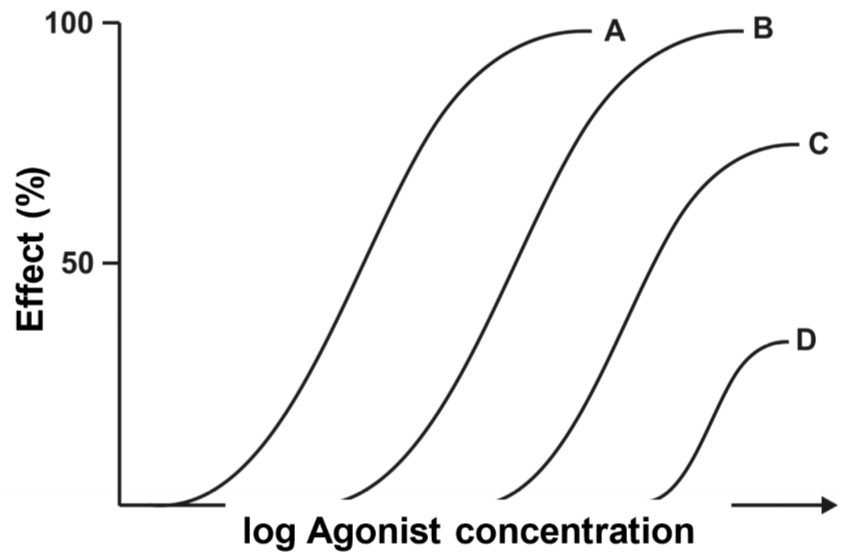
The drug-response curves looks different. The concept is the same, curve A was drawn with no antagonist present and curve D was drawn with the highest concentration of antagonist. The first thing we can see is that curve B has the same maximum effect; they both reach 100% effect. However, when the antagonist concentration is increased, the maximum effect starts to decrease. The maximum effect of C is approximately 70% and 30% for D.
So we must answer the following question: Why is the effect of the agonist not changed when a low concentration of antagonist is given?
To answer that we need to think back to topic 5, to the concept of receptor reserve. We found that most drugs can reach their maximum effect without occupying all the receptors, meaning that there is a “reserve” of receptors that don’t need to bind the drug to have full effect. As we saw in the drug-occupancy curves (two figures above), the occupancy of the agonist in case B was around 60%. This means that 40% of the receptors were occupied by the antagonist. However, in the drug-response curve we can see that in case B the agonist still reaches full effect. How come? It’s because the agonist has a receptor reserve of 40% or more! However, the receptor reserve is less than 60%, because in case C the maximum effect is much lower than 100%.
We say that this type of antagonism is surmountable as long as the antagonist occupies less receptors than the receptor reserve. The antagonism is insurmountable after that.
It’s important to note that irreversible antagonists are not widely used in clinical practice. This is mostly due to the fact that we cannot do anything is we give a too large dose or if we want to reverse the dose.
Non-competitive antagonisms
In the case of allosteric antagonism, the antagonist doesn’t bind to the same site as the agonist; it binds to an allosteric site. This decreases the effect of the receptor. Because the antagonist and agonist don’t bind to the same site, this type of antagonism is not competitive.
Another type of antagonism is “antagonism by inhibition of signal transduction”. Instead of the drug binding to the receptor and changing it somehow, the drug binds to a “downstream” protein involved in the signal transduction of the receptor. The drug can for example pass the cell membrane and inhibit a protein kinase that is usually activated by the receptor, like PKC or PKA. This type of antagonism is also not competitive.
Then we have functional (or physiological) antagonism. This happens when two different agonists bind to two different receptors, but the effect of the two receptors have opposing effects on the cell. For example, if both insulin and glucagon is given to a hepatocyte, the drugs will antagonize each other, because insulin wants to tell the hepatocyte to decrease gluconeogenesis, but glucagon wants to tell the cell to increase it.
We can also have antagonism by pharmacokinetic mechanisms. This means that instead of giving a drug that acts directly on the drug we want to antagonize, we give another drug that either inhibits the absorption of the first drug, enhances its breakdown or enhances its excretion. This type of antagonism is all about decreasing the concentration of the target drug in the patient, either by decreasing its absorption or by making sure it disappears from the body faster.
The last type of antagonism is chemical antagonism. This type of antagonist directly binds to the target drug to inhibit it. If the patient has gotten an overdose of heparin for example, we can give a drug called protamine which will bind to heparin and inactivate it.
Inverse agonism
Inverse agonists are a weird type of drug. When we give an inverse agonist to a tissue in vitro, that is, an isolated piece of tissue in the laboratory, the inverse agonist can decrease the activity of the receptor, doing the opposite of an agonist. However, this effect is only seen in vitro. When inverse agonists are given in vivo, that is, in a human body, they act just the same as competitive antagonists. In fact, many competitive antagonists used in clinical practice are actually inverse agonists.
Margin of safety
The concept of margin of safety was introduced in topic 5. We’ll talk more about it here.
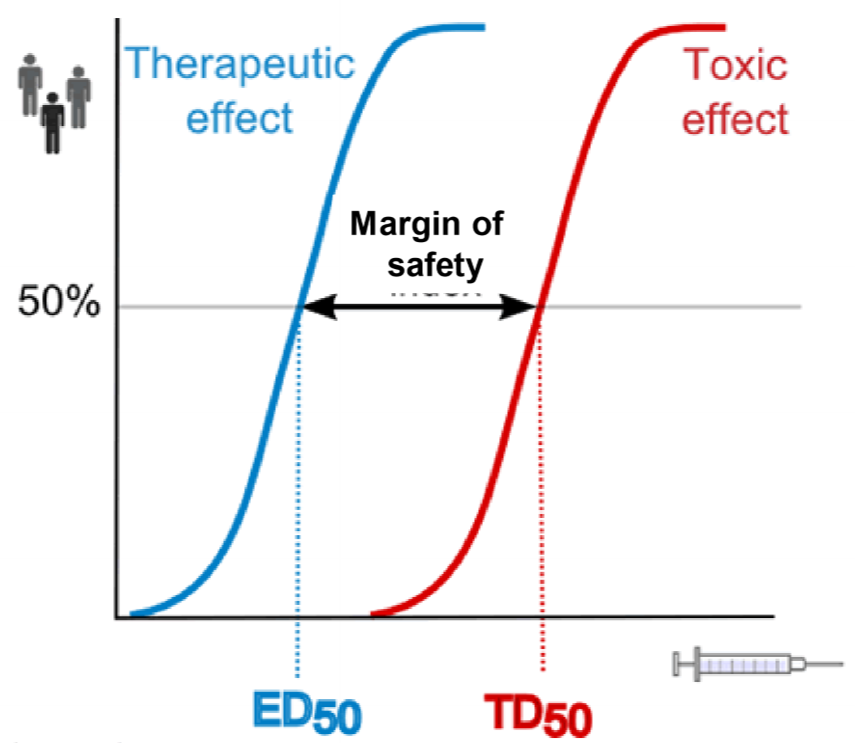
This figure shows what we mean by margin of safety. The two curves are for the same drug. To calculate the blue one we give a drug in many different doses and see how large of a fraction of patients are successfully treated by that dose. The dose that successfully treats 50% of the patients is called ED50. The red curve is calculated the same way, but instead of looking for the treating effect, we look for the toxic effects. TD50 is the drug dose that gives toxic effects in 50% of the patients tested.
The ratio of the two, TD50/ED50 gives the margin of safety, also called the therapeutic index. It shows how “far” away the toxic dose is from the therapeutic dose. For drugs like antihistamines the margin is very large, because the dose needed to give toxic effects is much larger than the dose needed for the clinical effect. However, for certain anticancer drugs, the margin of safety equals 1. This means that TD50 and ED50 are the same, so the drug dose that gives the therapeutic effect in 50% of patients also gives toxic effects in 50% of patients! This is because anticancer drugs are very toxic for the body, as the drug harms the body at the same time as it harms the cancer.
Why are inverse agonists not seen in vivo?
Good question. I don’t know.
Hello!
You mentioned that inverse agonist decrease the activity of receptor in vitro and act the same way as competitive antagonist in vivo, doesn’t that mean the same thing in both vivo and vitro then?
No, a competitive antagonist does not decrease the activity of the receptor in vivo. It simply competes with agonists, which decreases the stimulation of the receptor by the aforementioned agonists