Table of Contents
Page created on March 5, 2018. Last updated on June 26, 2020 at 12:11
Learning objectives
- What is the function of glycogen?
- Describe the structure of glycogen.
- What are the fates of glycogen in skeletal muscle?
- What are the fates of glycogen in the liver?
- Why is it an advantage that glycogen is branched?
- Which enzymes are involved in the synthesis of glycogen?
- Which enzymes are involved in the breakdown of glycogen?
General
Glycogen is the most important molecule for storing carbohydrates in humans. It’s a complex polymer, meaning that it consists of many repeating units. These units are molecules of glucose, making glycogen a polysaccharide.
Glycogen is found in largest amounts in the liver and in skeletal muscle, but is present in many other cell types, too, like the heart. Muscle contains approx. 250g of glycogen, while the liver contains approx. 75g.
In histology, glycogen (and mucus) is stained with the PAS stain, which stains the cytoplasm of highly glycogen-containing cells purple.
Glucose homeostasis
Glucose is the main energy source for most organs. Some organs, like the brain, can’t utilize alternative energy sources like fatty acids, so there should always be glucose in the blood. The concentration of blood glucose should always be > 3,5 mM.
One of the liver’s most important tasks is to maintain the level of glucose in the blood. After a meal the blood glucose level is very high, but it quickly normalizes as the tissues of the body take in the glucose, both for energy consumption and to store it in the form of glycogen. Between meals, during fasting, the liver must provide the blood with glucose. This glucose originates mostly from the breakdown of glycogen, but also from gluconeogenesis. The amount of glycogen in a liver can maintain the blood glucose level for 1 day of fasting – after this point the glycogen stores are depleted, and so glucose only originates from gluconeogenesis.
Skeletal muscle need a lot of energy while working, which is why this organ contains a lot of glycogen. Skeletal muscle doesn’t have glucose 6-phosphatase and therefore can’t release the stored glycogen into the blood as glucose, and therefore can’t help the liver maintain the blood glucose level. Instead, muscular glycogen provides energy for the muscles themselves. The liver does not used glycogen to provide energy for itself.
Structure
Glycogen is a large molecule, containing up to 50 000 units of glucose. The fourth carbon atom of the first unit of glucose is bound together with the first carbon atom of the glucose and so on, forming long chains of glucose molecules. These are called (α1 -> 4) glycosidic bonds.
The molecule is not a straight chain, but has branches. Every 8 – 12 unit of glucose an (α1 -> 6) glycosidic bond is formed, which creates a branch. Each branch can have more branches, creating a highly branched molecule.
Because it is branched, the glycogen molecule has many ends, however it only has one reducing end – all the other ends are non-reducing. Compare it to a tree, which only has one stem “end” but many leaf “ends”.
The advantage of these branches is that the number of non-reducing ends are increased from 1 to many. The enzymes which synthesize and break down glycogen can only act on non-reducing ends. By increasing the number of these ends, the enzymes can work at many ends simultaneously, which massively increases the speed of degradation and synthesis.
Each of these glycogen molecules form so-called β-particles. 20 – 40 β-particles form a so-called α-rosette, which can be visualized on electron microscopy.
Glycogenesis / glycogen synthesis
A protein called glycogenin is attached to the reducing end. It’s an enzyme which acts as a primer, polymerizing the first few glucose molecules.
After that another enzyme called glycogen synthase will catalyse the formation of further (α1 -> 4) glycosidic bonds as it attaches more and more glucose molecules to the glycogen. The pathway of synthesis goes as follows: glucose -> G6P -> G1P -> UDP-glucose. The glucose unit of UDP-glucose is then attached to a non-reducing end of glycogen by glycogen synthase, which releases free UDP.
Glycogen synthase can only catalyse the creation of (α1 -> 4) bonds. For the creation of the branches in the glycogen molecule, glycogen branching enzyme is needed. This enzymes forms the (α1 -> 6) bonds.
Glycogenolysis / glycogen degradation
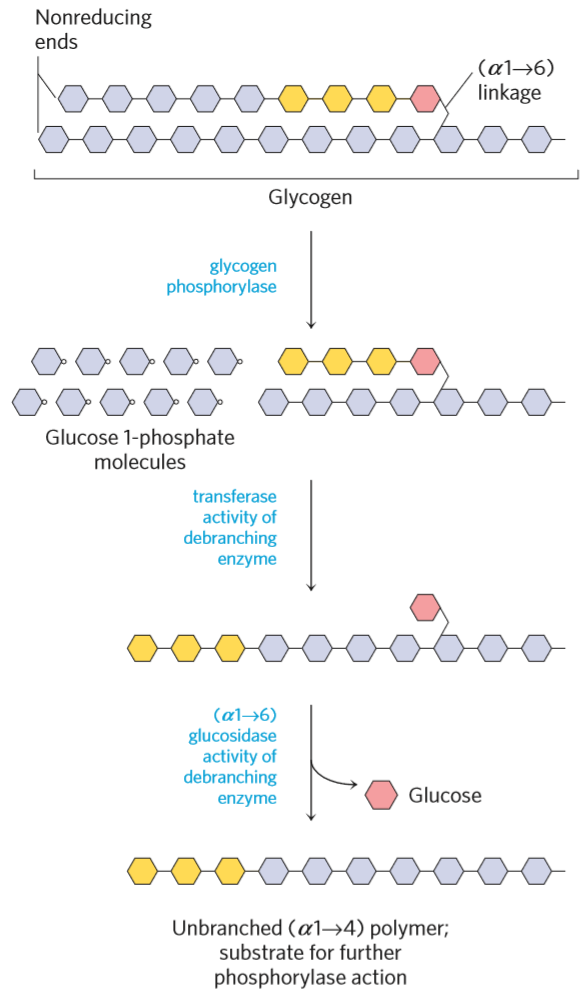
Degradation of glycogen. Each hexagon is a molecule of glucose.
Glycogen is degraded by glycogen phosphorylase and debranching enzyme. Glycogen phosphorylase converts the glucose units into glucose 1-phosphate by breaking of (α1 -> 4) bonds.
The debranching enzyme has two activities, transferase activity and glucosidase activity. When glycogen phosphorylase has reached the last 4 glucose units of a branch, the transferase activity of debranching enzyme takes the outermost 3 glucose units and puts them on the “main chain”, while leaving a branch of just 1 glucose. The glucosidase activity of debranching enzyme converts the last glucose on the branch into glucose (not glucose 1-phosphate!) by breaking the (α1 -> 6) bond.
Some glycogen is not degraded by glycogen phosphorylase and debranching enzymes, but rather in lysosomes by an enzyme called lysosomal α-glucosidase.
Glucose 1-phosphate can be converted into G6P, which can be further converted into glucose (in liver only), or go into the glycolysis (in muscle).
Regulation
Regulation is covered in the next chapter.
Glycogen storage diseases
Glycogen storage diseases are hereditary diseases caused by defects in enzymes related to glycogenolysis or glycogenesis. These defects prevent normal breakdown or synthesis of glycogen. There are many types, depending on the enzyme affected. These conditions generally cause fasting hypoglycaemia and enlarged liver.
Von Gierke disease occurs due to defect in glucose 6-phosphatase. Cori disease occurs due to defect in debranching enzyme. Pompe disease occurs due to a defect in lysosomal α-glucosidase. McArdle disease occurs due to a defect in the isoform of glycogen phosphorylase found in muscle.
Summary
- What is the function of glycogen?
- Glycogen acts as a storage molecule for glucose
- Describe the structure of glycogen.
- It’s a large molecule composed of up to 50 000 glucose units bound by α1 -> 4 bonds, with α1 -> 6 bonds every 8-12 residues to create branches.
- What are the fates of glycogen in skeletal muscle?
- Glucose 1-phosphate is released from glycogen, converted into glucose 6-phosphate and used in glycolysis
- What are the fates of glycogen in the liver?
- Glucose 1-phosphate is released from glycogen, converted into G6P and then into glucose to be released into the bloodstream
- Why is it an advantage that glycogen is branched?
- Synthesis and degradation enzymes can only act on the non-reducing ends, so having many of these ends speeds up the function of these enzymes
- Which enzymes are involved in the synthesis of glycogen?
- Glycogenin, glycogen synthase
- Which enzymes are involved in the breakdown of glycogen?
- Glycogen phosphorylase, debranching enzyme, lysosomal α-glucosidase
What is the difference between reducing end and non reducing end of glycogen?
The two ends of glycogen are different. one is reducing (can reduce other compounds) and one isn’t. To not confuse the two ends, one is named the reducing end and the other the non reducing end.
sorry for bothering you so much,
” How do we regenerate glycolytic NADH in aerobic and anaerobic condition?”
Aerobic: Nadh dehydrogenase
anaerobic: latate dehydrogenase
there are the answer from mari questions but i am not sure about it they use nadh rather than regenerate ,,
I don’t know the answer to that, unfortunately.