Page created on March 7, 2018. Last updated on November 19, 2018 at 17:16
Many intracellular signalling pathways are sensitive to reactive oxygen species, or ROS. The main sources of ROS in cardiac muscle cells are NO synthases, NADPH oxidases and mitochondria. Low levels of ROS may not be pathological and is necessary for some pathways, but high levels result in significant pathology. Mostly, ROS disturb posttranslational modifications of proteins involved in signalling, which disturbs the signalling.
The PARP pathway is important to understand the pathology (recall it from Biochemistry 1). ROS cause damage to DNA, and too large damage causes the cell to undergo apoptosis through the PARP pathway, which will cause tissue injury if it happens to many cells.
Mitochondrial dysfunction is the major source of ROS in cases where ROS levels are very high. ROS in turn will damage the mitochondria, which worsens the problem further. Ca2+ and ROS both induce the formation of a protein called mitochondrial permeability transition pore, or PTP, on the inner mitochondrial membrane. Should levels of Ca2+ and ROS stay pathologically high, the pore will open which will allow many proteins to enter the mitochondria that normally shouldn’t enter. This causes it to swell, which may cause the outer membrane to rupture. This releases cytochrome C, a protein in the respiratory chain, to leak into the cytoplasm, which causes the cell to undergo apoptosis.
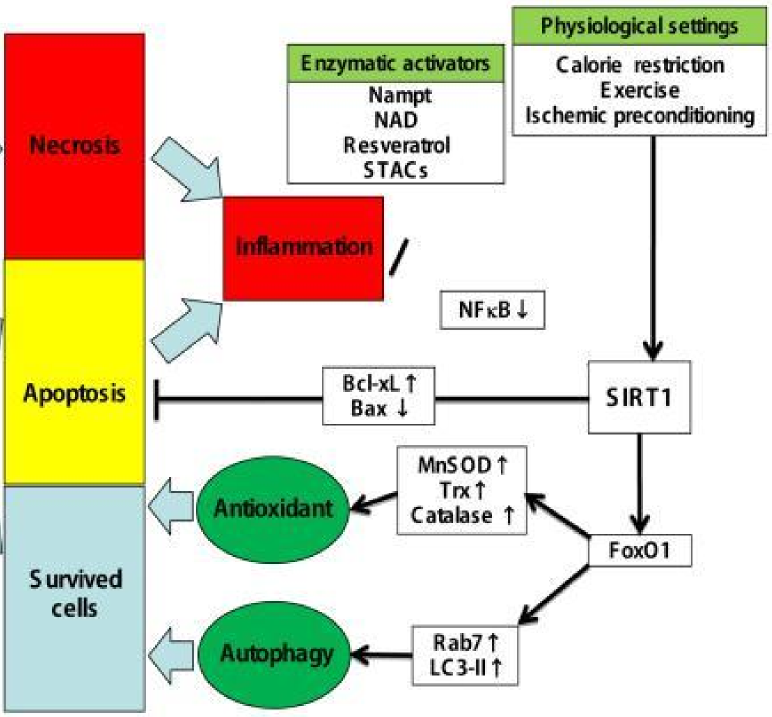
SIRT1
SIRT1 is a protein that protects the heart from damages during myocardial ischemia. SIRT1 is a deacetylase, so it activates and inactivates proteins by removing an acetyl from them.
The main function of SIRT1 is to suppress ROS production, apoptosis and inflammation. SIRT1 is activated by exercise and calorie restriction, meaning that exercising and reducing calorie intake will help a person survive myocardial ischemias.
SIRT1 activates the transcription factor FOXO1, which (among other things) increases transcription of enzymes that break down ROS, like catalase and SOD. SIRT1 also directly inactivates BAX while activating Bcl-2, which inhibits apoptosis.