Table of Contents
Page created on April 10, 2018. Last updated on November 19, 2018 at 17:16
Amyloids
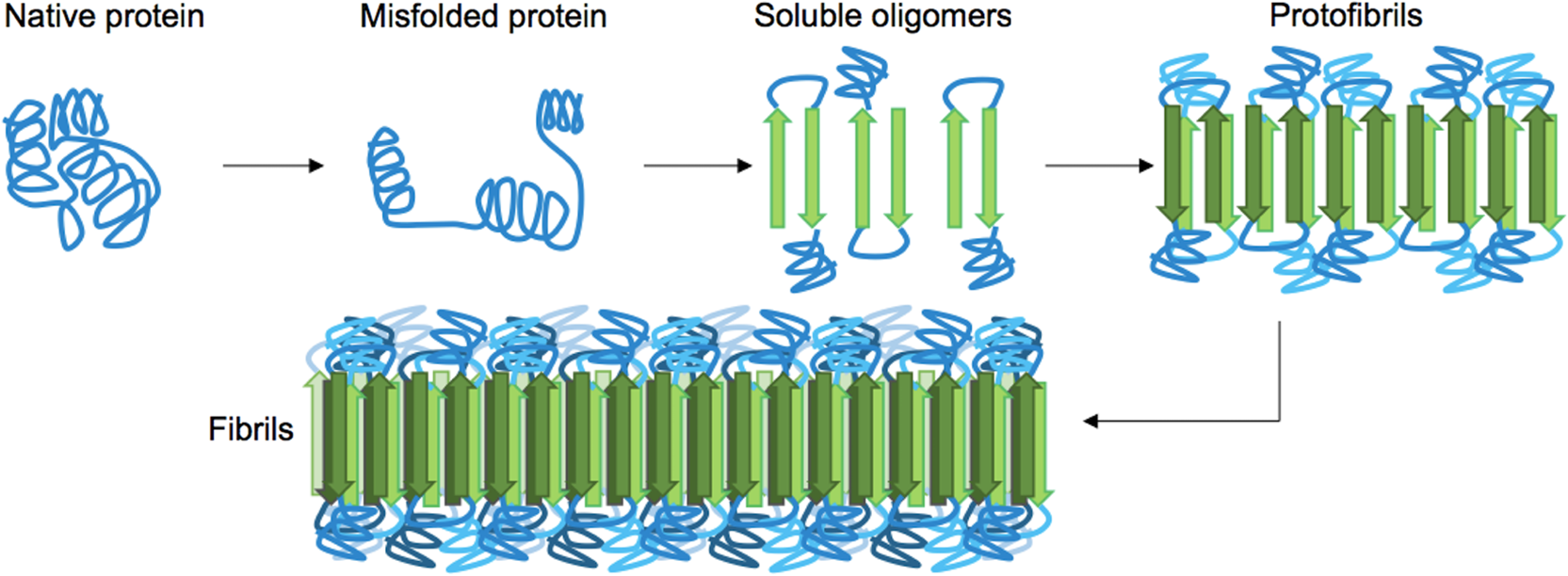
Amyloids are aggregates of proteins that have been folded into a special shape that allows the proteins to stick together to form fibrills. These amyloids are usually comprised of mostly β-sheets, so amyloids are sometimes called β-amyloids.
When proteins clump together in cells like this, the cells are damaged. Amyloid formation is pathological and doesn’t happen in healthy people. A disease where amyloid formation occurs is a type of amyloidosis. The specific protein that forms the amyloids vary from disease to disease: for example, in Alzheimers the amyloids are formed from a fragment of a protein called APP while in Parkinson’s, the protein α-synuclein forms the amyloids.
Alzheimer’s disease
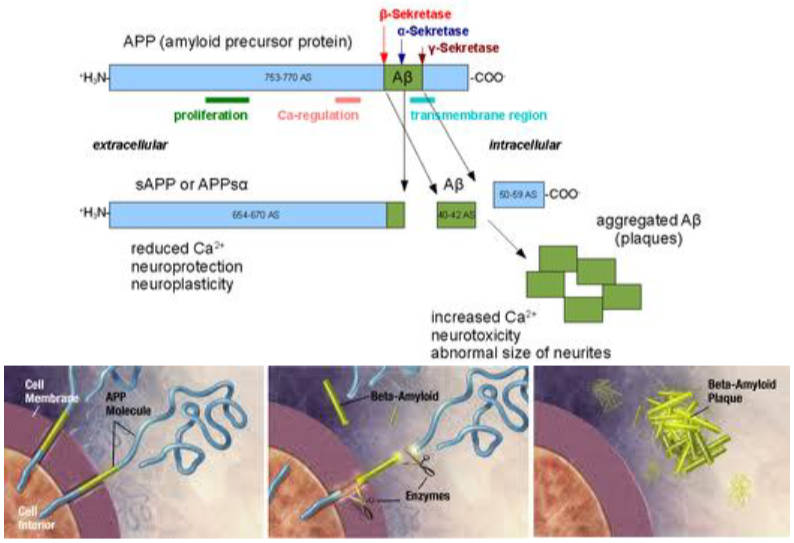
Alzheimer’s disease is, as mentioned, an amyloidosis. The fibrills formed in Alzheimer’s are called Aβ plaques and are the result of cleavage of a protein called APP at two specific sites. APP, or amyloid precursor protein, is a protein embedded in the cell membrane that is present in healthy people too. However, in some people is the protein cleaved by two enzymes called β-secretase and γ-secretase. This causes a fragment of the protein to be cleaved off, called Aβ. Many of these fragments will combine into the Aβ plaques that damage the cell.
In addition to amyloids, another type of molecule can cause Alzheimer’s: neurofibrillary tangles. Too much phosphorylation of a protein called tau is associated with many neurodegenerative diseases, together called taupathies. Alzheimer’s is one of them. Hyperphosphorylation of tau causes microtubules, a component of the cytoskeleton and extracellular matrix to be disintegrated. The hyperphosphorylates tau proteins aggregate and form the neurofibrillary tangles. You can read more about tau on topic 32.
Parkinson’s disease
Parkinson’s disease occurs when dopamin-producing cells in the substantia nigra in the brain die. When these cells die, they cannot stimulate cells in the striatum. The cells in the striatum will die from hypostimulation.
Many proteins and their genes are associated with Parkinson’s. Defect of either the protein or the gene can cause problems. Under pathological conditions, a protein called α-Synuclein can aggregate and form amyloids, which eventually forms what’s known as Lewy bodies, a classical histological marker of Parkinson’s.
Two proteins called Parkin and DJ-1 will, when mutated or defect, cause Parkinson’s by inhibiting ubiquitination and/or the proteasome. Two other proteins called PINK-1 and LRRK2 will, when mutated or defect, disrupt mitochondria and also cause Parkinson’s.
Huntington’s disease
Huntington’s disease is caused by loss of neurons in the caudate nucleus in the brain. It occurs when the gene which codes for a protein called Huntingtin, has too many CAG sequences in the gene. This occurs because of a special type of mutation called trinucleotide repeat expansion, which causes the trinucleotide CAG to repeat many times in a gene. Because of this is Huntington’s disease a type of trinucleotide expansion disorder.
Healthy people have less than 26 repeated CAG trinucleotides in the gene, but the higher the number above that, the higher the probability for a defective Huntingtin protein. CAG codes for the amino acid glutamine, so the result is a Huntingtin protein with too many glutamine residues. This causes the protein to be have a polyglutamine tract, causing the protein to be abnormal and damage other proteins in the neuronal cells.
Friedrieich’s ataxia
Friedrieich’s ataxia is also a trinucleotide expansion disorder, where the sequence GAA is repeated many times in the frataxin gene. This somehow reduces expression of this gene, causing too few copies of a protein called frataxin to be produced. The protein is important in formation of iron-sulphur clusters.
Prion disease
Prion diseases occur when special misfolded proteins gain the ability to cause other, correctly-folded proteins to misfold into non-functional proteins. This diseases are also a type of amyloidosis. In humans, the most important protein is PrP which is usually in the c-formation, called PrPC. However, this protein sometimes folds into the PrPSc form, which is resistant against proteolysis and therefore cannot be broken down.
When the PrPSc form of the protein comes into contact with another PrP protein in the PrPC form it will cause it to change into the PrPSc form. Many proteins in the PrPSc form will clump together and form amyloids which causes neurodegradation.