Page created on May 3, 2018. Last updated on November 19, 2018 at 17:16
Cyclin and CDKs are the proteins that control the cell cycle, and they’re highly regulated so that the cell division doesn’t proceed unless everything is cool with the cell. However, when this control fails, the cell can divide uncontrollably, which is what results in cancer. This almost always caused by mutations in multiple genes that is essential for the cell cycle regulation.
Oncogenes
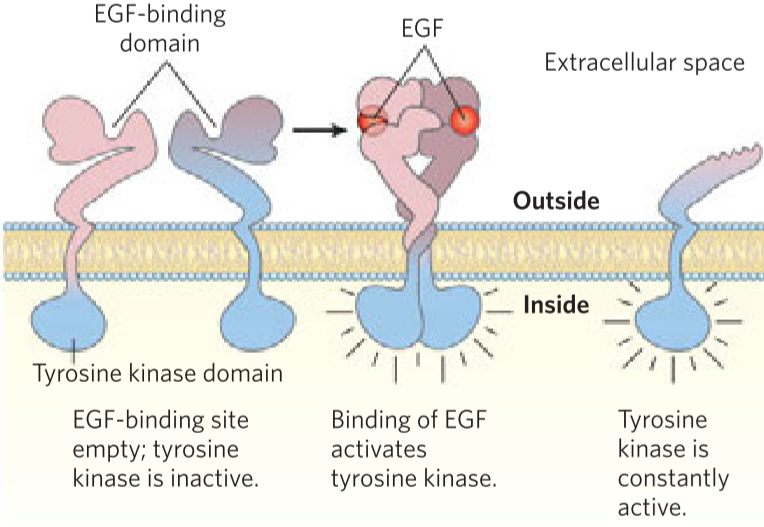
Proto-oncogenes are genes for normal, physiological proteins in the cell, but that have the potential to cause cancer if mutated. If these proto-oncogenes are mutated in a way that can cause cancer, they’re called oncogenes. The mutations that transform proto-oncogenes into oncogenes are called gain-of-function mutations, which means that when the gene is mutated, the cell will gain a new function, such as indefinite cell proliferation or increased protein expression. Gain-of-function mutations are dominant mutations, meaning that only one of the two copies of the gene needs to be mutated for the mutation to take effect. Examples are genes for growth factors, transcription factors or receptors.
One example is the receptor for the epidermal growth factor (EGF) called ErbB, which is normally present in many tissues without causing problems. The EGF normally signals to the cell that it should enter the S phase and start dividing. However, in many cancers, the gene for ErbB has been mutated and become an oncogene, which results in the ErbB receptor lacking a vital part. This mutated ErbB will send the “start replicating!” signal to the cell regardless of the presence of EGF. This will cause uncontrolled cell growth.
Small GTPases
There are two classes of G-proteins. The first is the familiar type which is involved in the G-protein coupled receptor pathways for example, like in glucagon or epinephrine pathway. The receptor gets activated, the G-protein binds a GTP instead of a GDP, and the G-protein will then activate other enzymes in the cell, like adenylyl cyclase. These G-proteins are actually large protein complexes comprised of multiple subunits that work together.
The other class of G-proteins are the small GTPases. They aren’t protein complexes but instead just small proteins that are attached to the membrane. Despite the name, they don’t just hydrolyze GTP; they have signalling functions as well. They’re not part of a receptor complex like the other type of G-proteins, but also transduce signals by turning on or off certain cellular processes. Like G-protein complexes, they are active when bound to GTP and inactive when bound to GDP. When it binds a GTP it becomes active, and will then activate other enzymes. After a while, the GTP will be hydrolyzed to GDP so the GTPase will be inactivated.
When the genes for the GTPases are mutated, the small GTPase proteins are defective as well. This can turn off the GTP hydrolysing activity of the protein. When this happens the GTP will never be hydrolyzed into GDP, which results in the signaling part of the small GTPase being active indefinitely! This causes whatever pathway the small GTPase is connected to to be activated without stopping. If the pathway in question is cell proliferation for example, the cell will continue to proliferate without stop, which can be cancerous.
When active, GTPases activate many different processes inside the cell. Recall that insulin activates cell proliferation. One of the GTPases, called Ras, is an essential part of the cell proliferation pathway by insulin. If Ras cannot be inactivated, the cell will continue to proliferate without stop, which can cause cause tumor formation. The gene for Ras is mutated in a considerable number of cancers, which makes the Ras gene a proto-oncogene.
Three other GTPases are important in cancer. The first is Rab, which is involved in transport of GLUT4 to the cell membrane and formation of transport vesicles around in the cell. The second is Ran which is involved in transport of proteins in and out of the nucleus. The third is Rho which is involved in the cell cycle.
Tumor suppressors
Mutations in proto-oncogenes can cause tumors, but mutations in tumor suppressor genes can do the same. Tumor suppressor proteins are proteins that ensure that the cell doesn’t replicate unless supposed to and only if there’s no DNA damage. In a way, tumor suppressor proteins are defense mechanisms against uncontrolled cell proliferation. However, if the gene for a tumor suppressor is mutated, this safeguard is removed, which can cause the cell to divide uncontrollably. This type of mutation is called a loss-of-function mutation, because the cell loses a function, such as the ability to regulate the cell cycle correctly. Loss-of-function mutations are recessive, meaning that both of the gene copies must be mutated for the mutation to be active.
Tumor suppressor genes and oncogenes are different in that aspect. Because there are two copies of each gene, the body often has a backup in case one of the genes are mutated. However, only one of the copies of the oncogenes need to be mutated to cause cancer. On the other hand, for mutations in tumor suppressor genes to cause cancer, both copies of the gene must be mutated.
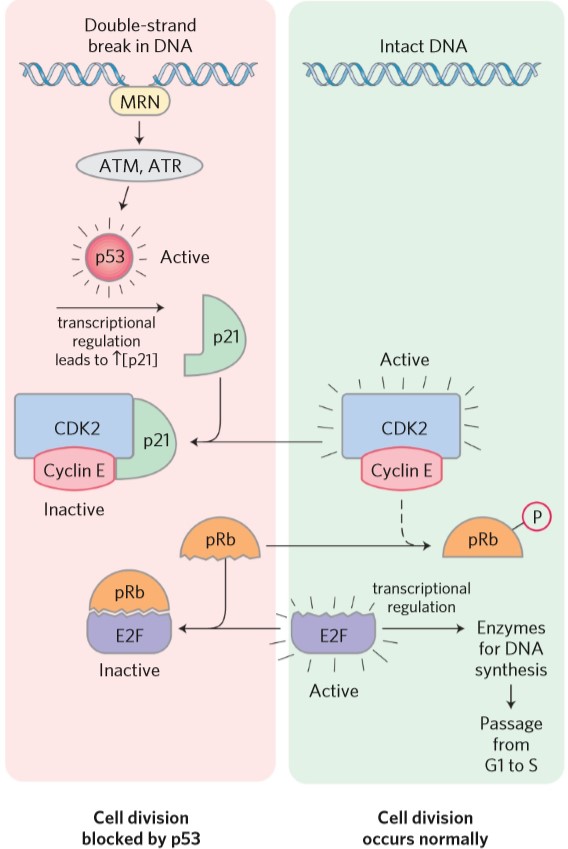
The important examples of tumor suppressors were mentioned in the “cell cycle” topic. Mutation in the gene for pRb will cause retinoblastoma, hence the name. Mutation in p53 can cause the cell to continue division despite having double-stranded DNA damage.
p21 is sometimes called cip and p27 is sometimes called kip. They are both part of the CIP/KIP family of CDK inhibitors.
We can think of mutations in proto-oncogenes as being the same as having the gas pedal in the car stuck at full speed, while mutations in tumor suppressor genes as being like having non-working breaks. Both will eventually cause a crash.
In practice, both oncogenes and defective tumor suppressors must be present for tumors to grow; it’s often not enough to just have an oncogene or just a defective tumor suppressor. This also works well with the car analogy.
The most important tumor suppressors are listed in this table.
| Name | Function |
| pRb | Inhibit E2F when double-stranded DNA damage is present |
| PTEN | Inhibits PDK1 and therefore PKB |
| p16 | Involved in senescence. |
| p21 (cip) | Inhibits CDK-cyclin heterodimer, inhibiting cell cycle |
| p27 (kip) | Inhibits CDK-cyclin heterodimer, inhibiting cell cycle |
| p53 | Transcription factor that increases transcription of p21 |