Table of Contents
Page created on October 7, 2019. Last updated on January 23, 2023 at 18:56
Haemostasis
Haemostasis is the body’s response to stop bleedings after endothelial damage. It consists of three steps:
- Primary haemostasis
- Local vasoconstriction
- Platelet plug formation
- Secondary haemostasis
- Clot formation
- Fibrinolysis
- Dissolving the blood clot
Coagulation factors
Most of the components involved in haemostasis are found in the blood. The first are the platelets. The second are the coagulation factors. The coagulation factors are proteins in the plasma that are involved in haemostasis. There are many coagulation factors. They travel in the blood in an inactive state. Most of them have two names, one which is just a roman numeral, and one which is their real name. Some coagulation factors are referred to by their roman number and some are referred to by their real name. Almost all of them are synthesized in the liver.
| Factor | Real name | Vitamin K required for synthesis |
| I | Fibrinogen | No |
| II | Prothrombin | Yes |
| III | Tissue factor | No |
| IV | Calcium ion (Ca2+) | No |
| V | (Not important) | No |
| VI | (Not important) | No |
| VII | (Not important) | Yes |
| VIII | (Not important) | No |
| IX | Thromboplastin | Yes |
| X | (Not important) | Yes |
| XI | (Not important) | No |
| XII | Hageman factor | No |
| XIII | Fibrin stabilizing factor | No |
| vWF | Von Willebrand factor | No |
| Prot. C | Protein C | Yes |
| Prot. S | Protein S | Yes |
| PKLK | Prekallikrein | No |
| HMWK | High molecular weight kallikrein | No |
The name that is usually preferred is bolded.
You should remember that factor II, VII, IX, X and protein C and protein S are vitamin K-dependent. This means that vitamin K is necessary for the liver to synthesize them. They’re also Ca2+ dependent in that they require Ca2+ to function.
Most coagulation factors are actually enzymes that can activate each other. They exist in an inactive state, but when one becomes activated, it can activate the others as well, forming a coagulation cascade, as we will see below.
A protein in an inactive state is usually given an “-ogen” suffix. As an example, the inactive form of plasmin is plasminogen, and the inactive form of fibrin is fibrinogen.
Primary haemostasis
When there is endothelial damage the first priority is to “plug” the hole. This is the goal of primary haemostasis.
Vasoconstriction:
The first component of primary haemostasis is vasoconstriction, the contraction of the smooth muscle in the wall of the broken vessel. This shrinks the lumen of the vessel, which decreases the blood flow through it. This makes it much easier for the plug to form and it decreases the amount of blood lost. This vasoconstriction is mediated by a local nervous pain reflex, and by the local release of vasoconstricting proteins like endothelin, one of the strongest vasoconstrictors.
Platelet plug formation:
The second component of primary haemostasis is the platelet plug formation. Formation of this plug requires platelet adherence, followed by platelet activation, then lastly by platelet aggregation.
When the endothelium of a vessel is damaged the blood will be exposed to the proteins that lie beneath the endothelium, especially collagen, which is abundant in the interstitium. Platelets in the blood recognize and adhere strongly to the collagen using the glycoprotein Ia/IIb receptor, both directly and with the help of von Willebrand factor (vWF). von Willebrand factor is released by injured endothelial cells and binds to exposed collagen, serving as binding sites for the platelet glycoprotein Ib receptor to bind to.
The adherence of platelets to collagen activates the platelets, causing them to release the content of their granules into the surrounding blood. These granules contain many compounds, like serotonin, vWF, ADP and thromboxane A2. These compounds further stimulate vasoconstriction of the vessel, and they stimulate other platelets nearby to become activated as well. ADP binds to receptors P2Y1 and P2Y12 to mediate these effects.
All activated platelets bind to and stick to the damaged endothelium and to each other, a process called aggregation. Platelets adhere to each other by binding the platelet surface receptor GPIb/IX/V complex to von Willebrand factor (VWF) in the subendothelial matrix, and by binding to fibrinogen using the glycoprotein IIb/IIIa receptor on the platelet surface. Their activation causes them to change shape from round to a star-like shape. This change in shape makes it easier for the platelets to aggregate and form a platelet plug, also called a white plug.
This platelet plug isn’t very stable; it can loosen easily. To ensure that the platelet plug is stable enough to remain until the endothelial damage has been repaired, we must “secure” the plug in place. This is where secondary haemostasis comes in.
Secondary haemostasis
Secondary haemostasis is the process of stabilizing the platelet plug by forming a mesh of a protein called fibrin. This process is complicated.
The main players in primary haemostasis were the platelets. The main players in secondary haemostasis are the coagulation factors. These factors activate each other in a complicated cascade called the coagulation cascade.
The coagulation cascade is (somewhat arbitrarily) divided into two main pathways; the extrinsic pathway and the intrinsic pathway, both of which end in the common pathway. The final point of the extrinsic and intrinsic pathways is the activated factor X, which is also the beginning of the common pathway.
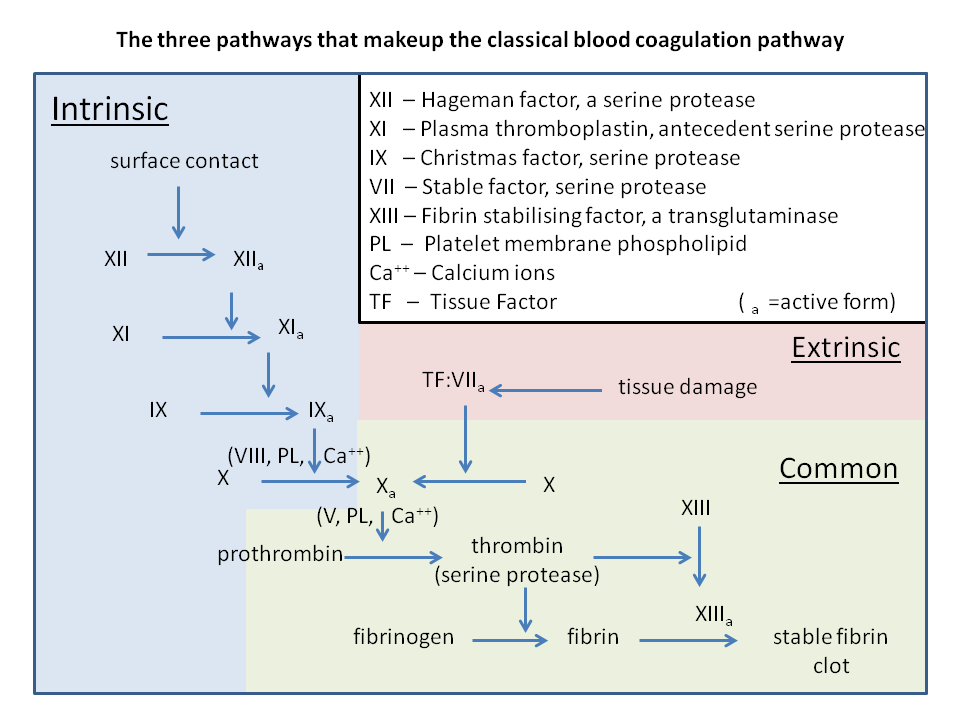
From https://en.wikipedia.org/wiki/Coagulation#Coagulation_cascade
Intrinsic pathway:
The intrinsic pathway, also called the contact activation pathway begins when the blood touches a negatively charged surface, such as collagen or glass. Glass is obviously not present in the body, but collagen is a major component of subendothelial tissues. As such, when endothelium is damaged, the blood is exposed to collagen, and so the intrinsic pathway initiates.
When blood touches the collagen or glass factor XII will be activated, turning into its activated form XIIa. Activated factor XII (XIIa) will activate factor XI. Activated factor XI (XIa) will activate factor IX. Activated factor IX (IXa) will, together with factor VIII activate factor X.
Extrinsic pathway:
A protein called tissue factor (TF) is also present in subendothelial tissue. When the endothelium of a vessel is damaged the underlying TF will be exposed to blood, which initiates the extrinsic pathway. Factor VII will bind to tissue factor and become activated. VIIa activates factor IX and factor X.
Common pathway:
Activated factor X (Xa) will activate factor II, converting it from prothrombin to thrombin. Thrombin will activate factor I, converting it from fibrinogen to fibrin. Thrombin also activates factor XIII.
Factor XIII binds to fibrin and forms crosslinks between fibrin monomers. This allows fibrin to polymerize and form a mesh that forms a “net” around the platelet plug and holds it in place. It is no longer a platelet plug but rather a blood clot or thrombus, and it is composed of fibrin, platelets and RBCs. It also contains plasminogen, which will be important in fibrinolysis.
The coagulation system is described as divided into the intrinsic and extrinsic pathways as a result of how the coagulation cascade acts in the laboratory (in vitro), not in the body (in vivo). The intrinsic pathway is initiated when blood comes in contact with glass, and the pathway was described according to what happened to the blood after this contact. The extrinsic pathway is initiated when blood comes in contact with thromboplastin, a mixutre of tissue factor and phospholipids. It’s actually suspected that the intrinsic pathway isn’t involved in haemostasis in the body at all, because deficiency of factors in this pathway doesn’t cause a defect in haemostasis (but rather in immunity).
Fibrinolysis
Fibrinolysis is the process where the blood clot is broken down. This process is activated simultaneously as the coagulation cascade, and both processes occur simultaneously. This is to prevent the blood clot from growing too large and occluding the whole blood vessel. Fibrinolysis is also involved in removing the blood clot completely after the damage to the blood vessel has been repaired.
Many proteins are involved in fibrinolysis:
- Plasminogen
- Activated form: plasmin
- Tissue plasminogen activator (tPA)
- Urokinase
- Factor XIIa
Plasminogen is an inactivated enzyme that travels with the blood. It can be activated by many proteins, like tissue plasminogen activator or urokinase. When activated it is converted into plasmin. Plasmin is an enzyme which dissolved fibrin, the main component of a blood clot.
The formed blood clot contains a lot of plasminogen. A few days after the blood clot has been formed and the bleeding has stopped will the endothelium secrete tissue plasminogen activator. This protein activates the plasminogen, which is contained in the blood clot, converting it into plasmin. Plasmin then dissolves the clot.
Organization
In some cases, the blood clots aren’t broken down by fibrinolysis but instead organized. This means that they’re slowly replaced by connective tissue formed by a type of cell called the fibroblast.
Anticoagulation
The body is constantly in a balance between procoagulation and anticoagulation. Both of these processes occur simultaneously and in a balance, so that neither too many (or too large) nor too few blood clots are formed. The term thrombosis refers to when a blood clot is too large and completely obstructs a blood vessel. This can be very dangerous and is the basis of heart attacks and strokes, among other diseases.
The body has many anticoagulant mechanisms:
- The surface of the endothelium is smooth, which prevents clots from accidentally forming
- Antithrombin – a protein produced by the liver which inhibits thrombin, IX, X, XI and XII
- Heparin – a molecule which stimulates antithrombin
- Protein C and protein S – proteins produced by the liver which inhibit factors V and VIII while stimulating plasmin formation
- Thrombomodulin – a protein produced by healthy endothelium which stimulates protein C
- Prostacyclin – a protein produced by healthy endothelium which inhibits platelet aggregation
Because multiple important coagulation factors depend on vitamin K to be synthesized correctly, a vitamin K deficiency will decrease the number of these coagulation factors. This causes excessive bleeding. A type of drugs called coumarins, especially warfarin, exploits this. Coumarins prevent the liver from utilizing vitamin K correctly, giving the same effects as vitamin K deficiency.
Coumarins are used to treat people who are at risk for thrombosis.
Coagulation tests
There are multiple tests we can perform on the blood to determine the function of the different parts of haemostasis.
The bleeding time determines the function of the vasoconstriction and platelet plug formation. It’s normally 2 – 4 minutes. It’s determined by puncturing the finger and measuring the time it takes for it to stop bleeding.
The clotting time determines the function of the intrinsic pathway. It’s normally 5 – 8 minutes. It’s determined by putting a drop of blood on a prewarmed piece of glass and checking how long it takes for a clot to form.
The prothrombin time determines the function of the extrinsic pathway and the vitamin K-dependent factors. It’s normally 15 – 20 seconds. It’s determined by seeing how long it takes for blood to clot after calcium and thromboplastin has been added to the blood.
The problem with prothrombin time is that the result varies significantly from lab to lab, depending on equipment and substrates used. To overcome this the result of the measurement needs to be normalized. This is done by inserting the prothrombin time into an equation, which gives you the international normalized ratio (INR). The INR is therefore a measurement of the prothrombin time, but it is standardized so that differences between laboratories doesn’t influence the result.
| Parameter | Function examined | Normal range | Elevated result in |
| Bleeding time | Primary haemostasis | 2 – 4 minutes | Thrombocytopaenia, von Willebrand disease |
| Clotting time | Intrinsic pathway | 5 – 8 minutes | Vitamin K deficiency, haemophilia |
| Prothrombin time | Extrinsic pathway, vitamin K-dependent factors | 15 – 20 seconds | Vitamin K deficiency, liver disease |
| INR | Extrinsic pathway, vitamin K-dependent factors | 1.0 | Vitamin K deficiency, liver disease |
Diseases of haemostasis
Many diseases of haemostasis exist, both congenital and acquired. Here are the most important ones and which parameter they affect:
| Disease | Pathological background | Pathway affected | Abnormal parameter |
| Thrombocytopaenia | Decreased number of platelets | Platelet plug formation | Elevated bleeding time |
| Von Willebrand disease | Deficiency of von Willebrand Factor | Platelet plug formation | Elevated bleeding time |
| Haemophilia A | Deficiency of factor VIII | Intrinsic pathway | Elevated clotting time |
| Haemophilia B | Deficiency of factor IX | Intrinsic pathway | Elevated clotting time |
| Haemophilia C | Deficiency of factor XI | Intrinsic pathway | Elevated clotting time |
| Vitamin K deficiency | Deficiency of vitamin K | Intrinsic and common pathways | Elevated clotting time, prothrombin time and INR |