Page created on October 7, 2018. Last updated on December 18, 2024 at 16:56
We’ve seen the haemoglobinogenic pigments, now it’s time to discuss the other endogenous pigments: lipofuscin, melanin and homogentisate.
Lipofuscin
We’ve met with lipofuscin before; it’s presence in normal in aged heart, liver and brain cells is physiological. The insoluble brownish-yellow “wear and tear” pigment is not dangerous to the cell but is a sign of earlier free radical injury.
The lipofuscin itself is a complex of lipids and proteins. The lipids used to be part of subcellular membranes, but these lipids were oxidized because of the presence of free radicals in the cell.
When lipofuscin is present in large amounts in a tissue is the appearance called “brown atrophy”.
Lipofuscin is seen in the recent infarct histoslide among others.
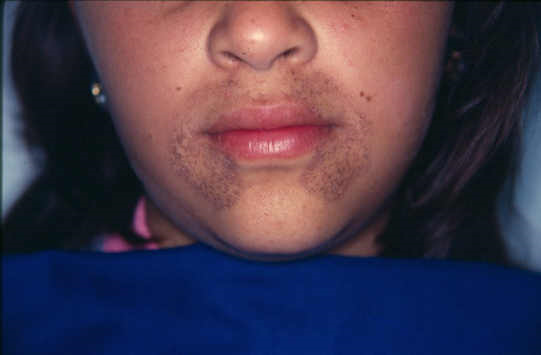
Melanin
Melanin is a more commonly seen pigment, as it’s present in (almost) everyone’s skin. It’s brown-black and is synthesized by melanocytes located in the epidermis. Its function is to protect us against ultraviolet radiation from the sun.
Although only melanocytes can produce melanin can it be found in adjacent keratinocytes in the skin and in dermal macrophages.
Homogentisate
Homogentisate, homogentisic acid or even alkapton is a breakdown product of tyrosine and phenylalanine, as you know from biochemistry.
The disease alkaptonuria is characterized by the presence of homogentisate in the urine, which gives it a brown or even black colour. Patients with this disease lack the enzyme homogentisate 1,2-dioxygenase so they can’t break down tyrosine or phenylalanine completely.
When homogentisate accumulates it polymerizes into a black pigment that can deposit in connective tissues, cartilages and joints. The condition where homogentisate has deposited in connective tissue is called ochronosis. Alkaptonuria is asymptomatic in the earlier years, but later the homogentisate deposits in the joints cause arthritis. It is an autosomal recessive condition.