Table of Contents
Page created on November 12, 2018. Last updated on November 15, 2020 at 12:51
Inflammation in general
Inflammation is the body’s protective response to remove the cause of cell injuries as well as eliminating damaged cells, necrotic cells and initiating regeneration.
The response involves host cells, blood vessels, proteins and other mediators.
What can cause inflammation? Some examples are:
- Infections
- Like common cold and pneumonia
- Allergies
- Hay fever and asthma
- Autoimmunity
- Thyroiditis, vasculitis and glomerulonephritis
- Chemical damage
- Alcoholic gastritis and hepatitis
- Physical damage
- Burn and radiation
- Heavy workout leading to repair of overused muscle fibres, and this makes you sore!
Inflammation plays also a role in diseases such as atherosclerosis, Alzheimer’s, prostate hyperplasia and malignant neoplasms.
Acute inflammation
An acute inflammation takes place minutes to hours after the “injury” and the cells involved are mostly neutrophil granulocytes. This leads to vascular changes like vasodilation and increased permeability, as well as increased adhesion and migration of leukocytes caused by activated endothelial cells. Let’s look at the changes in detail.
Vascular changes
At first, there will be a very short vasoconstriction due to axon reflexes activated by the sympathetic system when there is something damaging the tissue. Later, the release of histamine, NO and prostaglandins (PGI2) will cause arteriolar vasodilation. This vasodilation will result in hyperaemia, where the active hyperaemia causes increased arterial perfusion, while it passively gives congestion of the capillaries since the venous drainage is decreased.
The permeability of the vessels increases as well, giving leukocytes the opportunity to get to the site of inflammation. There are three responses causing increased permeability:
1. Immediate, transient response
The endothelium in postcapillary venules contract, caused by histamine, bradykinin and leukotrienes. When the endothelial cells contract will they open small spaces between themselves that allow cells to pass through.
This takes place within seconds and up to 15-10 minutes.
2. Immediate sustained response
The endothelial cells will suffer necrosis in arterioles, capillaries and venules at the site of the injury, where the cause for this is the direct injury.
This also happens within seconds but lasts for 4-6 hours.
3. Delayed, prolonged response
The endothelium of venules and capillaries will have cytoskeleton reorganisation and can undergo endothelial apoptosis. This happens 2-12 hours after injury and can last for hours or days.
Leukocyte-mediated endothelial damage can also increase permeability.
The increased permeability will lead to inflammatory oedema, where protein-rich fluid (exudate) suddenly can flow out of the vessels and will accumulate in the interstitial space.
The lymphatic vessels will also contribute in the inflammatory response. Lymph flow will be increased and helps drain oedema fluid, leukocytes and cell debris from the extracellular space. However, in severe inflammation, especially due to microbes, the lymphatics may transport the pathogens and become secondarily inflamed, a condition called lymphangitis. The draining lymph nodes can also get enlarged due to hyperplasia of lymphoid follicles.
Cellular events of acute inflammation
As you remember from physiology 1 and immunology, we’ve got a lot of different leukocytes in our body. In acute inflammations, the most important cell will be the neutrophil granulocyte, as it’s the most abundant leukocyte in our blood and therefore the first to accumulate.
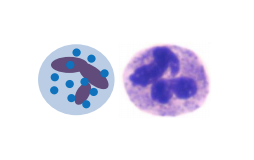
The neutrophil granulocyte has the following functions
- Phagocytosis
- Produces cytokines
- Produces antimicrobial agents
The first step is to recognize the potentially harmful agents, and this is done by the molecular pattern recognition, where the receptors on our cells can recognize structures that are common to many pathogens and dead cells. The most important families of these patterns are:
- Pathogen-associated molecular pattern (PAMP)
We have Toll-like receptors that recognizes e.g. Lipopolysaccharides (LPS) on Gram negative bacteria, endotoxins and flagellin. When the receptors bind to these kind of structures, NF-kB signalling gets activated.
- Danger-associated molecular pattern (DAMP)
NOD-like receptors will recognize e.g. ATP, uric acid associated proteins and extracellular matrix fragments which are products of dead cells. This leads to activation of inflammasome, a multi-protein cytoplasmic complex which in order activates Caspase-1. Caspase-1 cleaves the precursor of interleukin-1 into interleukin-1 (IL-1).
After the recognition of pathogens or dead cells, leukocytes must be recruited to the site of inflammation.
- Margination
First, margination will happen, where the leukocytes accumulate at the periphery of the vessels instead of in the centre due to stasis of the blood flow in the capillaries.
- Rolling
When the endothelial cells are already activated by local mediators and cytokines, they express adhesion molecules, allowing the leukocytes attach loosely to the surface and is able to “roll” on the vessel wall. This mechanism is called rolling, and the adhesion molecules are usually P-selectin and E-selectin.
- Adhesion
While the leukocyte is rolling on the vessel wall, it will search for integrins to adhere more strongly to the endothelial cells. The endothelial cells will express intercellular adhesion molecule-1 (ICAM1), that binds to Leukocyte function-associated antigen-1 (LFA-1) on the leukocyte, or Vascular cell adhesion molecule-1 (VCAM-1) that binds to integrin very late antigen-4 (VLA-4) on the leukocyte.
- Transmigration
After being arrested on the vessel wall, the leukocyte starts to migrate through the vessel wall by squeezing between intercellular junctions. Platelet endothelial cell adhesion molecule-1 (PECAM-1) is expressed on both leukocytes and endothelial cells to mediate the binding events of the leukocyte that are needed to cross the endothelium.
- Chemotaxis
After escaping the capillaries, the leukocyte moves along a chemical gradient toward the site of infection or injury.
When the leukocyte is at the inflammation site, it will get activated by microbes, products of necrotic cells and mediators. The removal of pathogens by phagocytosis follows these steps:
- Opsonisation
A type of proteins called opsonins, like the immunoglobulins, coat the surface of microbes and target them for phagocytosis. Leukocytes express receptors that bind to the opsonins, like Fc receptor for IgG (FcγRI), which can bind to IgG.
- Phagocytosis (engulfment)
The leukocytes’ membrane will zip around the microbe and create a phagosome inside the cell with the ingested microbe.
- Phagolysosome formation
Phagosome fuses with a lysosome, which contains digestive enzymes, forming a phagolysosome. The lysosomal enzymes digest the contents of the phagolysosome.
- Chemical killing
Rapid activation of leukocyte NADPH oxidase, also called phagocyte oxidase, converts oxygen into superoxide ion (O2–°), which will be converted spontaneously into H2O2. These ROS alone are not enough to kill all microbes, but lysosomes of neutrophils include the enzyme myeloperoxidase that converts H2O2 into hypochlorous radical (HOCl°), that can kill the remaining bacteria successfully.
Nitrogen derived free radicals act in the same way.
Extracellular killing
If the leukocyte fails to phagocytose whatever it wants to phagocytose can the toxic agents be released into the surroundings, which can damage the nearby cells. This is called frustrated phagocytosis. If a neutrophil tries to phagocytose immunocomplexes in the glomerular basement membrane can this happen, as the immunocomplexes are hard to phagocytose.
Some substances, like silica particles, can also rupture the membrane of phagolysosomes and leak the contents into the extracellular space.
Leukocytes also actively secrete granule components like elastase into the extracellular space, that digest and destroy microbes.
Extracellular traps are extracellular fibrillar networks of nuclear chromatin produced by neutrophil and eosinophil granulocytes in response to infectious pathogens, mainly bacteria and fungi. These traps contain antimicrobial substances and prevents spreading of microbes by trapping them. However, this leads to loss of the nuclei and the cell dies.
Mediators of inflammation
The mediators are usually produced at the site of inflammation by the cells there or are produced somewhere else but activated at the site of inflammation. They are tightly regulated and shortly lived.
We distinguish between cell derived mediators and plasma derived mediators.
Cell derived mediators
They are synthetized in response to stimulus and are rapidly secreted when the cell is activated. They are produced by either macrophage, mast cells, endothelial cells and leukocytes at the inflammation site.
| Mediator | Source | Actions |
| Histamine (vasoactive amine) | Mast cells, basophils and platelets | Vasodilation, increased permeability and endothelial activation |
| Serotonin (vasoactive amine) | Platelets | Both vasodilation and vasoconstriction, but mostly the former |
| Prostaglandins (arachidonic acid derivative) | Mast cells and leukocytes | Vasodilation, pain and fever |
| Leukotrienes (arachidonic acid derivative) | Mast cells and leukocytes | Increased permeability, leukocyte adhesion and activation |
| TNF-α | Macrophages, endothelial cells, dendritic cells, epithelial cells | Vasodilation, increased permeability, endothelial activation, decreased contractility of myocardium, cachexia, acute phase reaction |
| IL-1 | Macrophages, endothelial cells, dendritic cells, epithelial cells | Fever, pain, vasodilation |
| IL-6 | T-cell, macrophage, muscle cell, fat cell | Fever, acute phase reaction |
| Chemokines (cytokine) | Leukocytes and activated macrophages | Chemotaxis, leukocyte activation |
| Platelet activating factor (PAF) | Leukocytes and endothelial cells | Vasodilation, increased permeability, leukocyte adhesion, chemotaxis, degranulation and oxidative burst. |
| Nitrogen monoxide (NO) | Endothelium and macrophages | Vasodilation. Inhibition of both leukocyte-adhesion and platelet aggregation. |
Plasma protein derived mediators
These mediators circulate in their inactive form and undergo proteolytic cleavage upon activation at the inflammation site.
| Mediator | Source | Actions |
| Complement | Plasma, but produced in the liver | Leukocyte chemotaxis and activation. Direct target killing by MAC. Opsonisation. |
| Kinins | Plasma, but produced in liver | Increased permeability, vasodilation, pain. |
| Thrombin (clotting factor) | Plasma, but produced in liver | Leukocyte adhesion, PAF production, activation of arachidonate metabolism. |
| Plasmin (kinin) | Plasma, but produced in liver | Fibrin degradation, C3a conversion. |
“The endothelium in postcapillary venules contract, caused by histamine, bradykinin and leukotrienes.”
This is just a general question. I see that this is writtein in all other notes as well as yours, but I don’t really understand how histamine can cause the contraction?
I was hoping you could explain it perhaps? 🙂
Thanks in advance.
Hey!
Are you asking how histamine specifically can cause contraction? In that case, endothelial cells have histamine H1 receptors that histamine bind to that cause them to contract.
Or are you asking how endothelial cells contract? Because that appears to be very complicated, but I found a white-paper on the subject here: http://jcb.rupress.org/content/jcb/42/3/647.full.pdf
Thanks! That’s perfect 🙂
Hey (: under migration you then wrote margination happens first (cellular response to inflammation). so is the process of cells accumulating in the periphery of vessels called margination or migration?
Hey yourself! 🙂
It should’ve been margination. The process is called margination. Migration is another process where leukocytes migrate somewhere.
Fixed now, thanks!
in phagolysosome formation, don’t the enzymes digest the contents of the phagosome?
You’re right, I’ve updated the description to make that clearer.