Page created on November 22, 2018. Last updated on November 25, 2020 at 08:24
Autoimmune reactivity is in the background of many diseases, where the body reacts to proteins that should normally be present in the body, so-called self-antigens. Autoimmune diseases affect 2-5% of the developed country population and is increasing in incidence. These diseases vary in their extent, i.e. whether they affect many organs or cell types to cause systemic symptoms or just local symptoms.
Which part of the immune system of the 95% of the population is it that doesn’t work in people with autoimmune diseases? The keyword is immunologic tolerance.
Immunologic tolerance
Healthy, non-autoimmune immune systems shouldn’t react against self-antigen, they should tolerate them. Immunological tolerance or self-tolerance is this lack of immune response against self-antigens, and it is this property that people suffering from autoimmune disorders lack. The body implements many mechanisms to prevent immune cells from targeting self-antigens, and these mechanisms can be divided into two groups: central and peripheral tolerance.
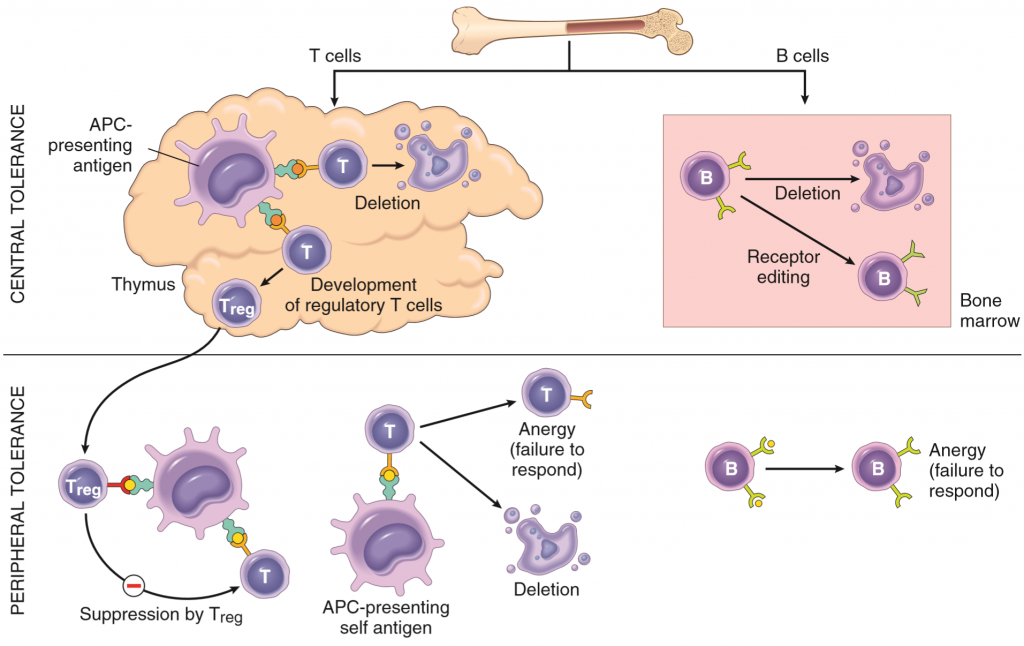
Central tolerance is the process where the body kills any T or B lymphocyte that would have reacted against self-antigens before they’re released into the circulation. This is done in the maturation process of both types of lymphocytes, which takes place in two different ways and places. T-cells are matured in the thymus. Thymic APCs continuously present self-antigens from the whole body to maturing T-cells with the help of a special transcription factor called AIRE. If any immature T-cell (called thymocyte) will recognize any of these self-antigens will the thymocyte be killed by apoptosis. This process is called negative selection. However, the thymic APCs cannot present every type of self-antigen in the body, meaning that the mature T-cells still react to some self-antigens. However, as we will see, this is usually not a problem.
B-cells mature in the bone marrow. They also undergo negative selection, similar to the thymocytes, to kill off any immature B-cell that would have developed into a self-reactive B-cell. Not all self-reactive B-cells are killed, some just have their receptors edited so that they’re no longer self-reactive. The negative selection of B-cells isn’t as strict as for the T-cells, meaning that self-reactive B-cells are regularly released into the circulation in small amounts. This is usually not a problem however, due to the peripheral tolerance.
Peripheral tolerance complements the central tolerance. Because central tolerance isn’t perfect is the peripheral tolerance essential to maintain immunological tolerance. The body has several mechanisms to “silence” or even kill peripheral self-reactive lymphocytes.
- Anergy refers to the functional inactivation of self-reactive lymphocytes. Recall from basic immunology that both B and T-cells need to be stimulated by TH cells or APCs respectively in order to become activated. If they don’t receive this stimulation will they not initiate the immune response. So, when a self-reactive lymphocyte recognizes a self-antigen and wants to become activated must it wait to be stimulated by other leukocytes. Because other leukocytes aren’t self-reactive as well will the self-reactive lymphocyte never receive this stimulation, so it won’t become activated.
- Regulatory T-cells are a special type of T-cells that suppress the activity of other leukocytes. They express the CD marker CD25, and have a special transcription factor called FoxP3, which is necessary for their development. Treg cells produce anti-inflammatory, immune-suppressive cytokines like IL-10 and TGF-β, and they block co-stimulatory molecules on APCs.
- Self-activation induced cell death is a mechanism where self-reacting lymphocytes are killed by apoptosis. This killing is mediated by the Fas–FasL pathway, which initiates the extrinsic pathway of apoptosis in the self-reacting lymphocyte.
- Sequestration of certain self-antigens from the lymphocytes can prevent that the lymphocyte reacts to them, simply because the lymphocytes will never meet with the antigens under normal circumstances. The word “sequestration” means isolated or hidden away, implying that some parts of the body are located in places where white blood cells can’t travel to, like the sperm in the testis (blood-testis barrier), brain (blood-brain barrier) and the eyeball. Because the immune cells can’t travel to those places can they not react against the self-antigens there. Certain intracellular proteins can also work as self-antigens, only provoking immune responses when necrosis causes the contents of the cell to be exposed.
Mechanisms of autoimmunity
Many genes and proteins are involved in maintaining immunological tolerance, and mutations in these genes can cause the tolerance to fail and an autoimmune disease to develop. Most autoimmune diseases can’t be explained by defects in single genes; most frequently are multiple gene mutations involved. Sometimes are also infections and tissue injuries involved.
Recall from immunology that the MHC I protein is coded by three genes: HLA-A, HLA-B and HLA-C. MHC II is coded by three other genes called HLA-DP, HLA-DQ and HLA-DR. Many variations (alleles) of these genes exists, and some alleles dramatically increase the risk of certain autoimmune diseases compared to other alleles. We often talk about the odds ratio, which is the risk of getting a certain disease when you have a certain allele of HLA. For example, the risk of developing ankylosing spondylitis is 100 times higher if you have a variant of HLA-B called HLA-B27, meaning that the odds ratio for HLA-B27 is 100. Autoimmunogenic HLA alleles are one variable that can contribute to autoimmunity. Below is a table of some diseases and the HLA alleles they are associated with. The table isn’t necessary to know by heart, but it might shed some light on the subject.
| Disease | HLA allele | Odds ratio |
| Ankylosing spondylitis | HLA-B27 | 100 |
| Coeliac disease | HLA-DQA10501 and HLA-DQB10201 | 7 |
| Multiple sclerosis | HLA-DRB11501 | 3 |
| Rheumatoid arthritis | HLA-DRB1 | 4-12 |
Mutations in some genes other than HLA are also important in certain autoimmune diseases, like:
| Gene | Function of gene product | Disease |
| PTPN-22 | Signalling in lymphocytes | Diabetes type 1, rheumatoid arthritis, inflammatory bowel disease |
| NOD-2 | Bacteria sensor in intestinal epithelial cells | Crohn’s disease |
| IL2RA | Part of the IL-2 receptor | Multiple sclerosis |
| FOXP3 | Transcription factor in Treg | IPEX |
| AIRE | Transcription factor in thymic APCs, important in negative selection | Autoimmune polyendocrine syndrome type 1 |
Infections may kickstart autoimmune diseases by many mechanisms.
- Molecular mimicry is a fancy word that describes how some microbes have epitopes that are very similar to self-antigens. When the body produces antibodies against these microbes will the antibodies also target the self-antigens, causing an immune response against them. The best example of this is a type II hypersensitivity called acute rheumatic fever, where antibodies produced against streptococci also target antigens on the myocardium, leading to myocarditis.
- Microbial infections cause APCs to upregulate their costimulatory molecules, in order to activate lymphocytes to fight the microbe. Recall that self-reactive T-cells become anergized because no APCs want to stimulate them. However, if this self-reactive T-cell by coincidence recognizes a self-antigen and gets stimulated by these APCs will the self-reactive T-cell be activated, starting an autoimmune response.
- Tissues damaged by infection can cause contents of cells to leak out and be exposed to the immune cells. These contents may act as self-antigens and initiate an autoimmune response
Autoimmune responses often promote further autoimmune responses, just like a ball that starts to roll and doesn’t stop. Tissue injury caused by the first autoimmune response may cause sequestered self-antigen inside cells to be exposed to the immune system, starting more autoimmune responses. This could be one of the mechanisms that cause autoimmune diseases to become chronic rather than acute.
I drew this topic on my exam and it came along with SLE, Rheumatoid Arthritis, Systemic Sclerosis and Sjörgen Syndrome.
So that’s a warning to future students
I am interested in using the figure on mechanisms of immunological tolerance in a text book I am writing. Do you know the original source?
Hello!
The original source is the book Robbins Basic Pathology. In the 9th edition it’s on page 122.
Good luck with your book!
Thank you
Hey,
I don’t understand the sequestration point you make 😔. Could you please explain. Just like the first sentence for that part.
Hey!
So some parts of the body have “barriers” that prevent white blood cells from entering these parts. Inside the testis are sperm cells maturating, and these sperm cells are different from the rest of the cells in the body, so the immune system would react to them. However, the blood-testis barrier, which is made by Sertoli cells, prevents white blood cells from ever coming in contact with the sperm cells. If they can’t come in contact with them and recognize them will the immune system never know that they exist and therefore not react against them.
The same principle goes for the blood-brain barrier and some parts of the eyeball. Both the brain and the eyeball have some proteins that the immune system could react against (autoimmune reaction), but the blood-brain barrier and some other barrier in the eye prevents white blood cells from ever coming in contact with these proteins.
I reworded that part of the topic as well to maybe make it more clear.
It would also be great to point out that sequestered actually means isolated or hidden away
The content of the cells may contain self-antigens, obviously not exposed to immune cells in normal conditions. If it is to leak however, they will cause an autoimmune response when exposed to immune cells.
Awesome topics btw!
Corrected the definition of “sequestered”.
Thank you!
Great work HABIBI , good luck with your exams .
Thank you, you too!