Table of Contents
Page created on November 22, 2018. Last updated on December 23, 2022 at 10:14
AIDS – Acquired Immunodeficiency Syndrome
AIDS is a retroviral disease caused by the human immunodeficiency virus (HIV). Its characterized by infection and depletion of CD4+ T-cells, and by severe immunosuppression which can lead to opportunistic infections, secondary neoplasms and neurologic manifestations that wouldn’t have happened in a healthy person.
Epidemiology
There are about 33 million people living with HIV worldwide, and the majority is in Africa. AIDS is the second leading cause of death in men between ages 25 and 44 years. Let’s look at the groups of people who has greater risk of getting it:
- Men who have sex with men
- Like Freddie Mercury
- Men who don’t have sex with men are also at risk but more frequently use protection, possibly because of pregnancy risks.
- HIV spreads more easily in the anus
- Intravenous drug abusers
- Sharing needles
- People with haemophilia
- Before 1985, they received factor concentrates from others’ blood.
- Recipients of blood and blood components
- If the blood giver has not been screened for blood borne infections properly, which is rarely a problem nowadays.
- Mother-child transmission
- HIV can travel through the placenta, infect during delivery or breast feeding.
Etiology
HIV belongs to the lentivirus family that is non-transforming and exists in two types; HIV-I and HIV-II. HIV-I is the most common type, while the second is pretty rare and is found in West Africa and India. Since HIV-II is so rare, we will consider only the pathogenesis of HIV-I.
Pathogenesis in the immune system
The major targets of HIV are the immune system and the CNS, but since we know more about how it attacks the immune system, we will discuss this in detail. We have to look at the structure of the virus. Its surrounded by a lipid envelope containing glycoproteins gp120 and gp41 which are attached to each other. These have a crucial role in the process of binding and entering into a host cell.
The virus is able to get through the cell membrane of CD4+ T-cells, macrophages and dendritic cells by binding to their CD4 molecule. However, that’s not enough to enter the host cell. HIV must also bind to the chemokine coreceptors, CCR5 or CXCR4 of the CD4+ cells! Note that the latter coreceptor is only found on T-cells.
The virus uses its gp120 to bind to CD4, which will make gp120 change its confirmation, giving it a new binding site. Gp120 then binds to the chemokine coreceptors, which induces gp41 to fuse with the cell membrane of the cell, making the virus able to enter the immune cell of the host!
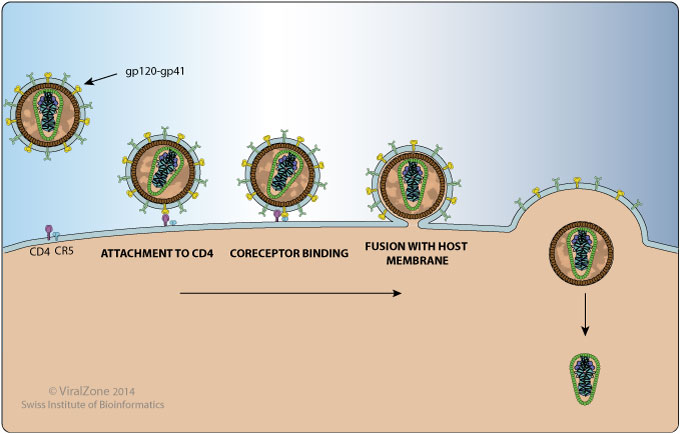
When inside the cell, the virus releases its reverse transcriptase, to transcript its RNA genome into the DNA of the host, making a complementary DNA, cDNA. In resting cells, the cDNA will be episomal, meaning that it will lay extrachromosomal, and not enter the genome of the host cell yet. However, as soon as the T-cell gets activated by cytokines or antigens, the viral DNA gets incorporated into the genome of the cell. The transcription of this cDNA will result in completely new HIV particles that bud from the cell membrane. The cell can actually fill up so much with HIV particles that the excessive budding kills it. The virus will spread further, however, repeating the process on new T-cells. Most of the times, the infection stays latent in the T-cells, and it can remain non-transcribed for months or years!
Like that isn’t enough, there are other causes for T-cell depletion in HIV. Chronic activation of uninfected T-cells by the HIV antigens makes them undergo apoptosis. Repeated infections lead to tissue destruction in lymphoid organs. Infected cells can bind to noninfected cells, creating syncytial giant cells, which makes the noninfected cells useless. Sneaky virus, right?
Pathogenesis in the central nervous system
As mentioned earlier, not much is known about HIV and CNS. However, we know that it targets monocytes and microglial cells as they are CD4+. The virus is carried into the brain by infected monocytes, and it believed that the neurons take damage from soluble factors produced by infected microglia, like IL-1, IL-6 and TNF. Neurons also take damage from NO induced by gp41 on the HIV envelope, but the neurons themselves never get infected since they lack the CD4 molecule!
Clinical course of the HIV infection
1. Acute retroviral infection
This stage is in the very beginning. Infected T-cells die on the mucosal surfaces. The immune system of the host develops a response against this virus, while the viral replication takes place in the lymph nodes. This leads to flu-like symptoms:
- Sore throat
- Myalgias
- Fever
- Weight loss
- Fatique
2. Middle, chronic phase
This stage is usually asymptomatic. The lymph nodes and the spleen will have continuous HIV replication and more T-cells die. In this phase, minor opportunistic infections may appear, like oral and vaginal candidiasis, herpes zoster and tuberculosis.
3. Clinical AIDS
The host defence is broken down, and the number of CD4+ T cells is minimal in blood, while plasma virus levels are high! If no treatment is given, the patient in this stage will not live longer than 7-10 years.
Now, more severe opportunistic infections can develop since the immune system barely works in the host. Usually, they are just latent since the body of a patient with HIV can’t get rid of them, and in this stage, they are enough to kill them. The infections can be:
- Pneumonia
- Bronchopneumonia killed Freddie Mercury
- Candidiasis, especially oral (fungal infection)
- Colitis
- Tuberculosis, both classical and avium
- Encephalitis by toxoplasma gondii
- Salmonella
AIDS and tumors
25-40 % of the untreated patients can develop tumours due to AIDS. Oncogenic DNA viruses like HPV and EBV, that are latent elsewise, get the opportunity to proliferate.
Kaposi sarcoma is a vascular tumour that is pretty rare in healthy persons, will become aggressive in patients with AIDS. B-cell lymphomas can also appear.
Therapy for AIDS
There is no cure for AIDS, however, there is medication for slowing down its progress. Antiretroviral drugs target the viral reverse transcriptase and integrase. Medication improve decrease the death rate dramatically as well as they decrease the incidences of pneumocystis and Kaposi sarcoma.
Transplantation immunity
The biggest problem of today’s organ transplantations is the immunological rejection by the body because it recognizes the transplanted organ as foreign. This rejection process can be both cell- or antibody-mediated. The transplanted organ is also called an allograft, or simply a graft.
Recognition of allografts
The host body recognizes that the transplanted organ is foreign mainly due to MHC molecules. Recall that there is a large variety in MHC molecules between people. In fact, it’s very unlikely for anyone other than monozygotic twins to have the exact same MHC molecules. This recognition takes place by two mechanisms, direct and indirect recognition.
Direct recognition occurs when host TC and TH cells recognize MHC I and MHC II on the surface of graft cells, respectively. This activates the host T-cells. TH cells initiate the immune response against the graft by producing cytokines, while the TC cells differentiate into cytotoxic T lymphocytes (CTLs) which start to kill the graft cell directly. The response by the TH cells is similar to a type IV hypersensitivity reaction.
Indirect recognition occurs after APCs have phagocytosed, processed and presented the processed pieces of graft MHC molecules on their own MHC II molecules. This process is the same that occurs when APCs meet microbes: Phagocytose, process, present on MHC II. TH cells then recognize the presented graft MHC molecules and become activated, inducing as in the direct recognition.
Recall that B cells also are APCs. When B cells phagocytose, process and present graft MHC molecules to TH cells will the B cells be stimulated to produce antibodies against the graft.
Effector mechanisms of graft rejection
As noted above are TC cells, TH cells and antibodies activated and targeting the graft at this point. The damage to the graft endothelium, causing ischaemia and thrombosis, is the most important aspect of graft rejection.
CTLs then proceed to kill graft cells. When CTLs kill graft endothelial cells will thrombosis and ischaemia of the graft occur. TH cells secrete cytokines which mediate a type IV hypersensitivity-like reaction in the graft tissue and blood vessels. TH cells also activate macrophages and lymphocytes, which also kills endothelial cells and cause ischaemia of the graft.
The antibodies produced against the graft are also important. They bind directly to MHC molecules on the surface of graft cells, especially on the graft endothelium, which recruits leukocytes to the endothelium and also activates the complement system.
Types of rejection
The hyperacute rejection occurs only in patients who already had antibodies against foreign MHC in their blood. Anti-MHC antibodies can be found in patients who have already rejected a transplant, in multiparous women (they form anti-paternal MHC molecules during pregnancy) or in patients who have previously received blood transfusion. In patients with anti-MHC antibodies will a new graft be rejected immediately, within minutes or hours. This occurs because the circulating antibodies rapidly bind to the endothelium of the grafted organ, which results in ischaemia and thrombosis.
Hyperacute rejection of a kidney transplant causes the kidney to become cyanotic, spotted, and produce no urine. Histologically we can see inflammation of the arteries and arterioles, thrombosis and ischaemic and fibrinoid necrosis.
The acute rejection occurs within weeks in patients who aren’t properly immunosuppressed after the transplant, and it can occur even in properly immunosuppressed people after years. It’s caused by both cellular and humoral (antibody) mechanisms, so we examine them separately.
Acute cellular rejection is most commonly seen within the first months. Histology shows extensive TC and TH infiltration of the transplanted organ, oedema and haemorrhage. The tubules are injured and there is endotheliitis. T-cells destroy the graft by destroying the graft parenchyme and vessels by cytotoxicity (TC) and inflammation (TH).
Acute humoral rejection mainly targets the vasculature, and causes necrotizing vasculitis, deposition of antibody, complement factors, fibrin and thrombosis in the vessels. Cells in the vessels may proliferate, causing the lumen to narrow which causes infarction or atrophy.
Chronic rejection manifests itself as a progressive, slow appearance of kidney failure, which can be measured by the level of creatinine in the blood. Morphologically the kidney parenchyme is being replaced by interstitial fibrosis. Proliferation of smooth muscle in the wall of arteries causes narrowing of the lumen with resulting hypoperfusion and ischaemia. It’s probably caused by T-cells producing cytokines that act on the smooth muscle cells.
Transplantation of haematopoietic stem cells
Some diseases require transplantation of haematopoietic stem cell, like aplastic anaemias, haematological malignancies, immunodeficiency states and severe thalassaemias. These stem cells can be taken by from bone marrow of the donor, from umbilical blood of new-borns, or by administering haematopoietic growth factors to healthy adults.
The recipient receives chemotherapy and/or radiation to kill malignant cells and to prepare the body for receiving transplanted stem cells. The stem cells are then given intravenously, where they find their way to the bone marrow and create their home there.
Rejection can occur in this type of transplantation as well. Rejection is probably mediated by T cells and NK cells that are resistant to radiation therapy and chemotherapy.
Graft-versus-host disease (GVHD) occurs when transplanted T cells (or T cell precursors) recognize their new body as “foreign” and react against it. TC and TH cells become activated, creating inflammation and killing host cells. GVHD may occur not only in haematopoietic stem cell transplant but also transplantation of solid organs which is rich in lymphoid cells, like the liver. By making sure that the donor and recipients’ HLA is similar (HLA typing), we can reduce the risk of GVHD.
Acute GVHD occurs within weeks of the transplantation and causes epithelial cell necrosis in three organs: liver, skin and the GI tract. The symptoms are jaundice, rash and diarrhoea, respectively.
Chronic GVHD can follow acute GVHD or appear later after a slow progression. It has symptoms similar to autoimmune disorders. Chronic liver disease, involution of lymphoid organs and recurrence of infection, potentially life-threatening ones, is characteristic.
T cells mediate GVHD, so a possible solution could be to deplete T cells in the donor before the transplant. This does reduce the risk for GVHD but increases the risk of recurrence of leukaemia (which is often treated with stem cell transplant).
Hey !! If we pick this topic on the exam , Do we need to tell this whole bunch of transplantation and rejection things?
By the way , I really appreciate your notes and works
<3
Glad you like my notes!
It’s hard for me to know exactly what you need to talk about and not on the exam, as I’m not an examiner. But I think I based the topic on the lecture, so one would assume that they’d expect at least some of it.
“Heterosexuals are better at using protection,” just sounds very discriminating. As a homosexual, I take safe sex very seriously and this just sounds like homosexuals are being looked down upon; heterosexuals are capable of being just as equally unsafe when it comes to sex…
Stating statistical facts are not discriminatory. It’s a known fact that, unfortunately, men who have sex with men less consistently use condoms compared to men who have sex with women. See for example this source https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3676274/
I am really glad you take safe sex seriously, but this article isn’t about you. And of course men who have sex with women are capable of being equally unsafe as men who have sex with men. I’ve never said otherwise.
Sorry to hear that the article disappointed you. I’ve changed the wording to sound less derogatory.
The article did not disappoint me, in fact I was enlightened more than anything really. Anyhow, thank you for the changes.
<3
“HIV spreads more easily in the anus, apparently.”
Logically, due to the lack of natural production of lubrication, and therefore the increased risk of tear during anal sex 🙂
Removed the “apparently” to reduce ambiguity