Page created on November 25, 2018. Last updated on December 13, 2018 at 14:48
Tumor suppressors
The genes that code for the proteins that create the checkpoints in the cell cycle are tumor suppressors. They work as brakes in the cell proliferation. The G1-S checkpoint checks the DNA for damage while the G2-M checkpoint quality checks the DNA after replication and determines whether it’s safe for the cell to divide. When cells sense DNA damage will the cell cycle stop at the checkpoint until the DNA is repaired, or the cell decides that the damage is so large that it should undergo apoptosis, or the cell will enter a senescent state, where it doesn’t replicate.
When the checkpoint proteins fail can the cell continue to replicate despite having accumulated DNA damage, which can promote cancer growth.
Retinoblastoma gene
Retinoblastoma gene, or RB, codes for a protein called pRb. It was one of the first tumor suppressors to be described and gets its name from the fact that it was first discovered in retinoblastoma tumor cells.
pRb can be phosphorylated. When its phosphorylation is decreased (hypophosphorylation) is the protein active, and when it is hyperphosphorylated is it inactive. Activated pRb controls the G1-S checkpoint by inhibiting the elongation factor E2F. Doing this blocks transcription of cyclin E, stopping the cell cycle.
Growth factors like EGF (epidermal growth factor) and PDGF (platelet-derived growth factor) induces phosphorylation of pRb, inactivating it and allowing the cell to continue with the cell cycle.
Recall that for a tumor suppressor to be inactivated must both alleles for it be defective. This means that people who only have one defect allele are perfectly healthy, however they’re obviously at a much higher risk to develop cancer, as they only need one spontaneous mutation to have a defective tumor suppressor instead of two.
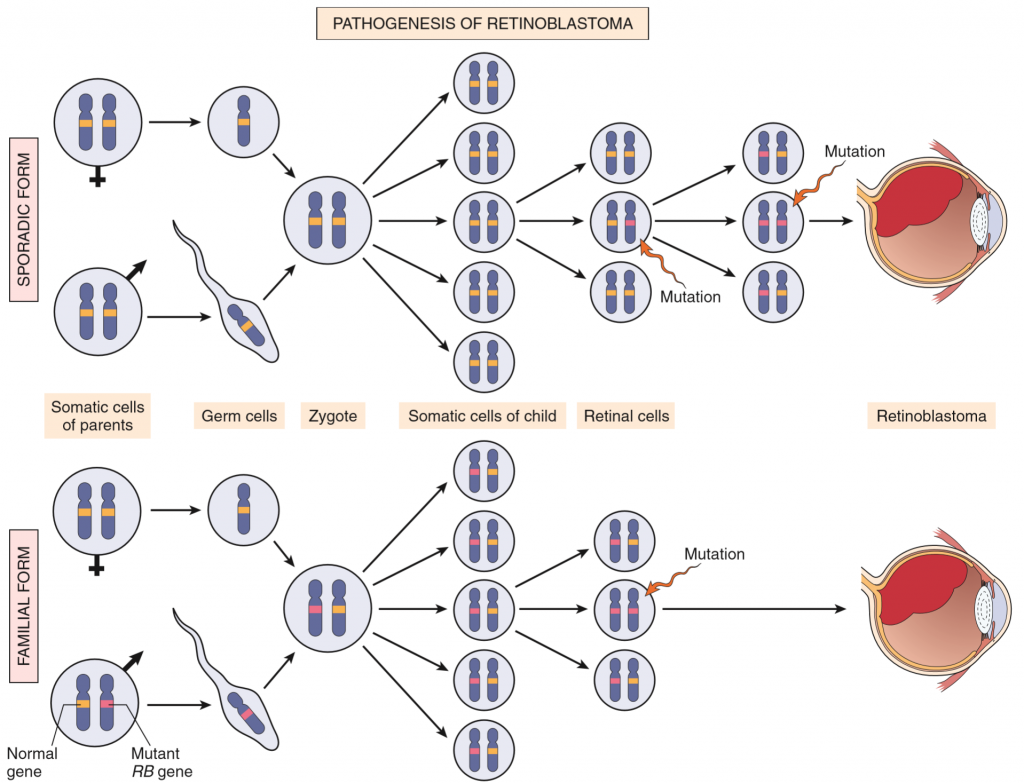
This figure illustrates that. The patient on the bottom has a germ-line mutation in one of the RB alleles, meaning that it only needs one spontaneous mutation in order to develop retinoblastoma. The patient on the top doesn’t have this germ-line mutation, so they must accumulate two spontaneous mutations to develop the cancer.
Some oncogenic viruses get their oncogenicity from interfering with pRb, like HPV, adenovirus and polyoma virus. These viruses produce proteins that bind to the active form of the protein, inactivating it.
This is a good point to introduce the two-hit hypothesis. For tumor suppressors like RB to lose their function must both alleles be mutated. In a normal person must therefore two mutations (two hits) occur to develop e.g. retinoblastoma. In families that carry one germ-line mutation of RB will children inherit this mutation. They already have one mutations, so they only need one mutation (one hit) to develop retinoblastoma.
RB mutations occur very, very frequently. They’re so frequent that, if you inherit a germ-line mutation in one RB gene, the chance that you’ll acquire the second hit (one more mutation) some time during your life is 90%! So not everyone who inherits the germ-line RB mutation will get retinoblastoma (10% won’t), but enough people do that we can say that retinoblastoma follows an autosomal dominant inheritance pattern.
p53
As introduced earlier is p53 known as the guardian of the genome, due to its importance in detecting DNA damage. p53 is activated by DNA damage or hypoxia, and causes the cell to either:
- Arrests the cell cycle in G1 to fix the damage. If that fails it will either:
- Undergo apoptosis or
- Become senescent (non-replicating)
p53 is a nuclear protein with very short half-life, meaning that it is very short-lived. p53 can’t be detected by immunohistochemistry in healthy cells, but it can in cells with mutated p53, because the mutated p53 accumulates in the nucleus.
Over 50% of human tumors contain p53 mutation. Like for RB is p53 only inactivated when both its alleles have been mutated. Also like for RB can this occur with two somatic mutations or one germ-line and one somatic mutation. Patients with the germ-line mutation have something called Li-Fraumeni syndrome, where they have a 25x greater chance of developing many malignant tumors, like sarcomas, breast cancer, leukaemia, brain tumors and adrenal cortex carcinomas.
Like for pRb can some viral proteins bind to and inactivate p53, like HPV viral protein E6.
Hey, Nik
Good notes btw,
just wanted to emphasize that RB is an autosomal dominant disease. However, the inheritance only effects one of the genes, not two as it must. The result is a heterozygous child. The clue in categorizing this as an AD disease is due to the fact that over 90% of these kids undergo a mutation, and thereafter develop RB.
I think this is a question some “malignant” professors might ask for a 5 on this topic.
Thank you for the kind words.
I didn’t know this, but now I do and have added it to the topic. Thanks!