Table of Contents
Page created on September 24, 2018. Last updated on December 4, 2020 at 16:47
Atherosclerosis means “gruel hardening” and is one of the worst guys when it comes to increased morbidity and mortality in the Western world. It underlies the pathogenesis of coronary, cerebral and peripheral vascular disease. So, make sure to understand and learn this well, because this will follow you through your whole career as a medical doctor. Also, don’t mix arteriosclerosis and atherosclerosis.
Thickening and hardening of the arterial wall is called arteriosclerosis. There are three general patterns of arteriosclerosis with different clinical and pathologic consequences:
- Atherosclerosis
- Mönckeberg medial sclerosis
- Arteriolosclerosis
We will discuss mostly atherosclerosis.
Arteriolosclerosis
Arteriolosclerosis affects small arteries and arterioles and may cause downstream ischemic injury. It’s related to hypertension.
Möckenberg medial sclerosis
This is a calcification of the walls of muscular arteries. It is typical of long-standing diabetes. Unlike the other types, this type does not narrow the vessel lumen (does not cause arteriolar stenosis), so it doesn’t cause disease. It usually occurs on the legs.
However, these arteries are so calcified that they can’t be compressed, which interferes with blood pressure measurement, and makes it seem like the blood pressure through the arteries is much better than it actually is. This is especially important in people with peripheral artery disease and type 2 diabetes, as in these patients the so-called ankle-brachial index (ABI) can’t be used to estimate blood flow in the leg.
Atherosclerosis
Its characterized by the presence of intimal lesions called atherosclerotic plaques which are composed of soft lumpy lipid cores, mainly cholesterol and cholesteryl esters with some necrotic debris, covered by a fibrous cap. These plaques can mechanically obstruct the vascular lumen, and if they rupture it can result in a catastrophic vessel thrombosis. They also tend to weaken the underlying vessel tunica media, making them prone to aneurysm formation.
The risk factors for atherosclerosis are:
- Age
- Male gender
- Hyperlipidemia
- Bad diet
- Bad lifestyle
- Hypertension
- Smoking
- Diabetes
- Inflammation
Pathogenesis
There are two theories about the pathogenesis of AS, one emphasizing the response to endothelial injury, while the other focuses on repeated formation and organization of thrombi. Both Robbins and the lecture focuses most on the response to injury theory.
This model views atherosclerosis as a chronic inflammatory disease and a problem with the healing response of the arterial wall to endothelial injury. Lesion progression involves interaction of modified lipoproteins, macrophages and T lymphocytes with endothelial and smooth muscle cells of the arterial wall. Let’s take a closer look at how this atherogenesis takes place, step by step.
- Endothelial injury
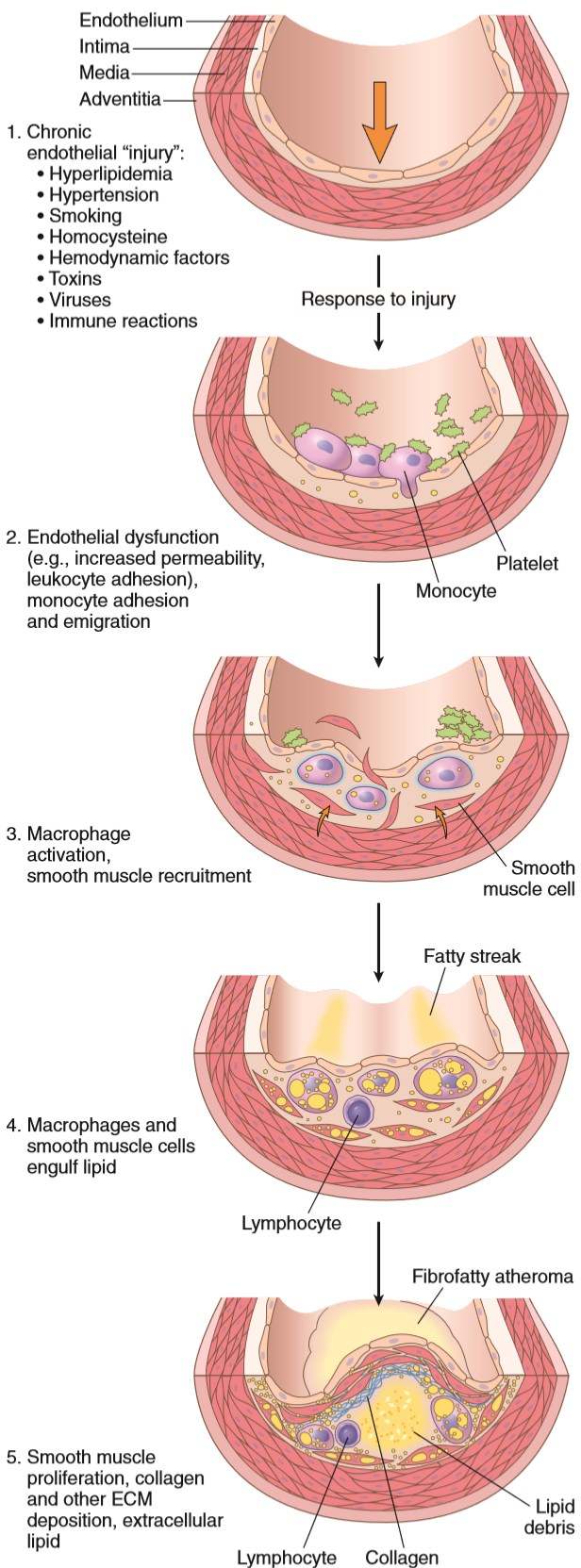 This is the cornerstone of the hypothesis. Injury to endothelium results in thickening of the tunica intima. However, this endothelium will still be intact, but dysfunctional. These dysfunctional cells will have increased permeability, increased leukocyte adhesion and altered gene expression, that are important contributors to atherosclerosis.
This is the cornerstone of the hypothesis. Injury to endothelium results in thickening of the tunica intima. However, this endothelium will still be intact, but dysfunctional. These dysfunctional cells will have increased permeability, increased leukocyte adhesion and altered gene expression, that are important contributors to atherosclerosis.
The two most important causes of endothelial dysfunction are hemodynamic disturbances and hypercholesterolemia.
- Hemodynamic disturbances
The plaques tend to occur at the openings of exiting vessels, at branching points and along the posterior wall of the abdominal aorta. What do they have in common? They are the sites of turbulent blood flow.
- Lipid accumulation in vessel wall
It’s not a surprise that cholesterol and cholesteryl esters are the dominant lipids when it comes to atherosclerotic plaques. Chronic hyperlipidemia leads to an accumulation of lipoproteins in the tunica intima, where they generate two pathogenic derivatives, oxidized LDL and cholesterol crystals. The oxidized LDL comes from LDL that has been oxidized by free radicals due to impaired endothelial cell function. LDL is then ingested by macrophages through their scavenger receptor (receptors on macrophages and other cells that take out pathogens). This causes the macrophages to become full of fat, and at this point we call them foam cells. The oxidized LDL stimulates also local release of growth factor, cytokines and chemokines which will increase monocyte recruitment. This molecule itself is cytotoxic to endothelium and smooth muscle cells as well.
- Inflammation
As we know, normal and healthy vessels do not bind inflammatory cells. But as soon there is an injury, as in early atherogenesis, the dysfunctional endothelial cells will start to express adhesion molecules, especially vascular cell adhesion molecule-1 (VCAM-1), to bind monocytes and T-cells.
The monocytes recruited to the tunica intima will differentiate into macrophages and engulf the lipoproteins accumulated there, including the oxidized LDL and small cholesterol crystals. However, activated macrophages produce reactive oxygen species that will continue to oxidize LDL and stimulate smooth muscle cell proliferation.
T-cells will interact with the macrophages and secrete inflammatory cytokines (e.g. IFN-y) which stimulate the macrophages, endothelial cells and smooth muscle cells, making the inflammation chronic.
- Infection
There is circumstantial evidence linking infections as Herpes virus, cytomegalovirus and Chlamydia pneumoniae have been found in atherosclerotic plaque. However, these infections are as common as atherosclerosis, making it hard to draw conclusions. Also, atherosclerosis has been induced in germ-free mice.
- Smooth muscle proliferation and matrix synthesis
The proliferation of smooth muscle cells and extracellular matrix (ECM) leads to conversion of the earliest lesion, a fatty streak, into a mature plaque. The intimal smooth muscle cells can originate from the tunica media or from circulating precursors, but either way they will synthetize ECM at the site at the atherogenesis, using the newly synthetized ECM to stabilize the plaques. If there are activated inflammatory cells at the site, they will induce apoptosis of smooth muscle cells and breakdown of ECM, which may cause the plaque to become unstable.
Morphology
This topic has still not come to an end… There’s still morphology and complications left. So, let’s take a look at the morphology.
- Fatty streaks
Appear as yellow, flat macules that merge into elongated lesions, 1 cm in length or more. They are composed of lipid-filled foamy macrophages, and do not cause any disturbance for the blood flow. Fatty streaks may physiologically appear in aortas of infants and are present in all children older than 10 years. Fatty streaks may evolve into plaques, but not all are destined to progress.
- Atherosclerotic plaques
White to yellow raised patchy lesions that can range from 0,3 to 1,5 cm in diameter but can sometimes be bigger. They are often found at one portion of any given arterial wall where the blood flow is turbulent.
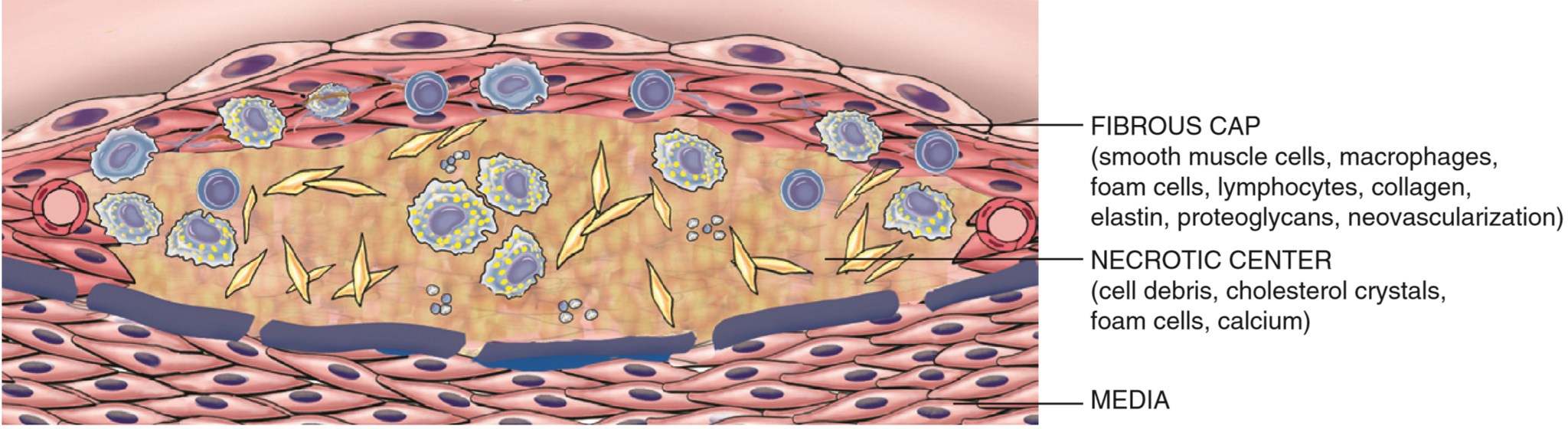
The plaques have three principal components:
- Cells, like smooth muscle cells, macrophages and T-cells.
- Extracellular matrix including collagen, elastic fibers and proteoglycans
- Intracellular and extracellular lipids.
The plaques have a superficial fibrous cap composed of smooth muscle cells and dense collagen. Under it we can find a necrotic core containing lipids as cholesterol and cholesteryl esters, necrotic debris, foam cells, fibrin and other plasma proteins. They also often become calcified.
Clinical consequences
Now it’s finally time for clinical consequences. You’re soon done with this topic!
- Atherosclerotic stenosis
If the plaque is stable, clinical consequences usually present when the artery lumen is 70% occluded. In the coronaries, when the 70 % of the artery is occluded, the patient will have enough cardiac perfusion when in rest, but with even the mildest exertion the demands of oxygen will exceed the supply. This will lead to chest pain because of cardiac ischemia, also called stable angina.
- Acute plaque erosion typically triggers thrombosis, because the exposed plaque is a good surface for blood to clot on. This can lead to the lumen becoming even more occluded, leading to either partial or complete obstruction and often tissue infarction.
- Plaque ruptures can expose atherosclerotic debris and lead to distal embolization, which can become catastrophic.
- The plaque also destroys the underlying vessel wall, which can lead to aneurysm.
- Bleeding into the plaque (from below) expands the volume of the plaque, making it occlude the vessel even more.
first of all, thank you for these awesome notes ! lifesaver
there is a typos in the first part. as arteriosclerosis is the general term for hardening of a wall which have 3 patterns : 1. atherosclerosis 2. arterioLOsclerosis (not arteriosclerosis) 3. monckenberg medial sclerosis. then the next paragraph (arteriolosclerosis instead of arteriosclerosis)should be fixed too
Thank you! I’ve corrected the errors.
SO if i say in the exam that Chrisk is the most important risk factor of atherosclerosis will I pass ?
100% guaranteed.
Nice Article