Table of Contents
Page created on March 16, 2019. Last updated on April 14, 2022 at 15:42
Introduction
We’re entering the topics about haematological malignancies. Before we start, we need to make a few concepts clear first.
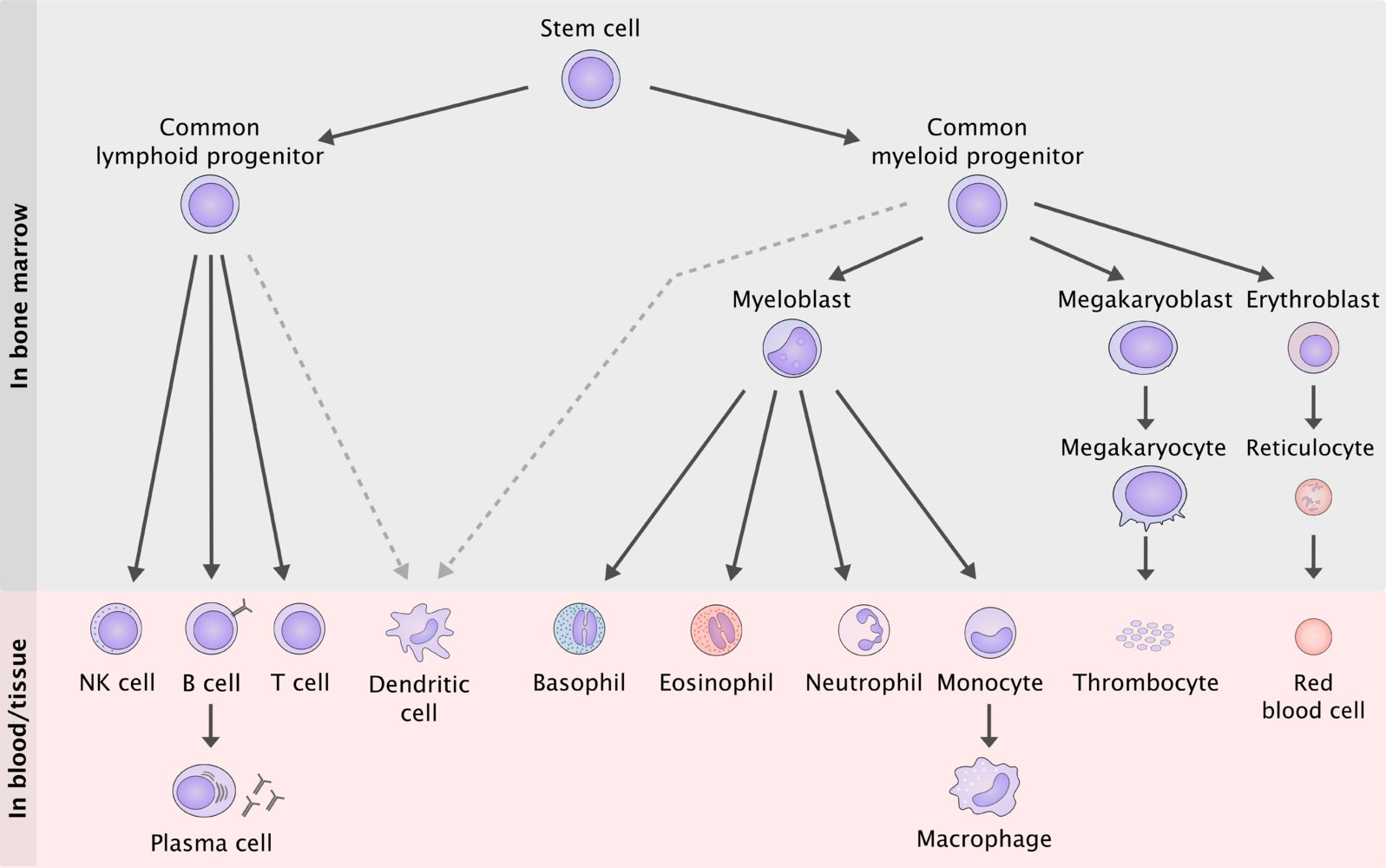
The figure above shows the normal haematopoiesis. Based on this figure can we divide the white blood cells into two categories: The myeloid cells, which includes the cell lines on the right, and the lymphoid cells, which includes the cell lines on the left.
There are three major types of haematological malignancies:
- Lymphoid neoplasms – where cancer cells originate from T cells, B cells or NK cells
- Myeloid neoplasms – where cancer cells originate from myeloid progenitor cells
- Histiocytic/dendritic neoplasms – where cancer cells originate from macrophages or dendritic cells
Lymphoid neoplasms may manifest as leukaemias, in which the tumor cells are primarily present in the peripheral blood and bone marrow, or as lymphomas, in which the tumors cells primarily produce masses in lymph nodes or other tissues.
Myeloid neoplasms may affect either of the three myeloid cell lines, erythroid (RBCs), granulocytic (WBCs except lymphocytes), or megakaryocytic (thrombocytes). This may result in elevated count of one or more of these types of cells in the blood. If WBCs are elevated, there is myeloid leukaemia.
The different neoplasms are named according to their usual manifestation. If a certain neoplasm always or almost always forms a lymphoid mass it is called a lymphoma, like Burkitt lymphoma. If a certain neoplasm always or almost always causes tumor cells to be present in the blood is it called a leukaemia, like acute lymphoblastic leukaemia. However, there is often some overlap.
Proliferating tumor cells in the bone marrow displace and destroy the normal cells of the bone marrow, which can cause bone marrow failure with decreased proliferation of the other cell lines, potantially causing anaemia, thrombocytopaenia, and/or neutropenia. This can occur in both lymphoid and myeloid neoplasms.
Lymphoid neoplasms
Classification
The lymphoid neoplasms are classified according to multiple properties:
- Whether they are Hodgkin lymphoma or non-Hodgkin lymphoma
- Whether they originate from T or NK cells or from B cells
- Whether they are aggressive, indolent (not so aggressive) or in-between (“other”)
- Whether they originate from precursor cells (which don’t express CD20) or mature cells (which express CD20)
The classification is best visualized like this:
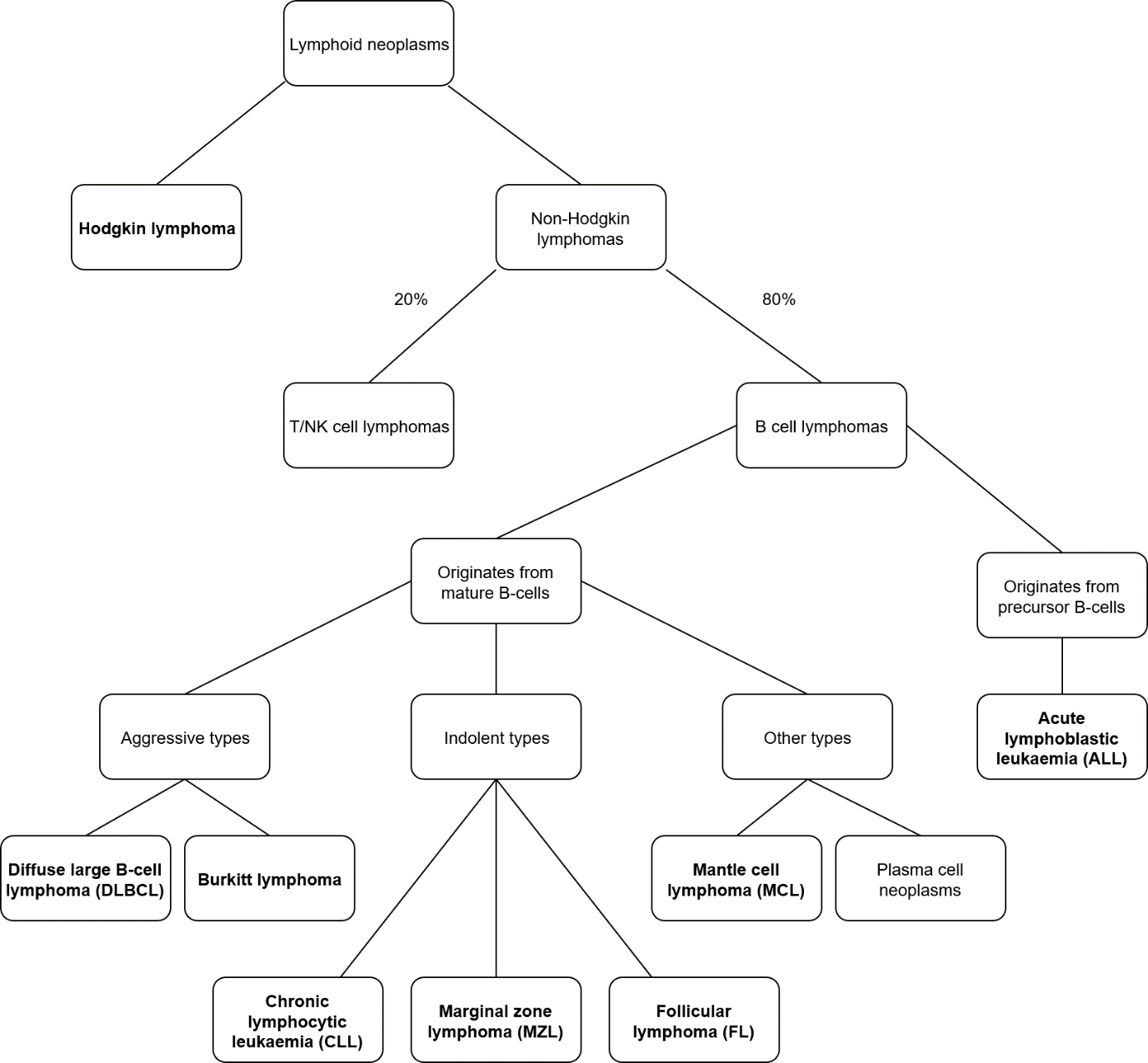
The bolded boxes are specific names of neoplasms. The non-bolded boxes show the classifications. This list is not conclusive.
As you can see from the figure above, there are many lymphoid neoplasms that are classified under “non-Hodgkin lymphoma”. Most (80%) of these originate from B-cells. Hodgkin lymphoma always originates from B-cells and is covered in a different topic.
Lymphomas may be nodal or extranodal, where “nodal” refers to a lymph node. A lymphoma is nodal if it originates in a lymph node, and primary extranodal if it starts in other lymphoid tissues, like the lymphoid tissues in the GI tract, the skin or in the CNS. A lymphoma can be “secondary” extranodal if it spreads from a lymph node to other tissues.
Diffuse large B-cell lymphoma is the most common form of non-Hodgkin lymphoma, followed by follicular lymphoma.
Clinical features
Lymphoid neoplasms that affect lymph nodes cause painless and firm lymphadenomegaly. Infiltration of tumor cells into the spleen and liver can cause hepato and/or -splenomegaly as well.
The term B symptoms refer to systemic symptoms of fever, DRENCHING night sweats, and unintentional weight loss, which are common symptoms in patients with lymphoid neoplasms, more often in aggressive ones than indolent ones. Other common symptoms include lymphadenomegaly and splenomegaly.
Aggressive B-cell lymphomas
Aggressive lymphomas are very aggressive malignancies, as they proliferate rapidly and untreated the life expectancy is just a few weeks. They can be considered “high-grade”. However, thanks to their rapid growth, they are potentially curable. They are more frequent in children.
Indolent B-cell lymphomas
Indolent lymphomas are much less harmful than the aggressive ones, so they can be considered “low-grade”. In many cases, patients die with indolent lymphomas rather than of them. They remain asymptomatic for many years, and are therefore often diagnosed incidentally on a laboratory test rather than due to the patient presenting with symptoms.
In some cases they may not even require treatment, as they progress so slowly. Even untreated, the life expectancy is multiple years. The tumor cells don’t proliferate fast. In most cases, indolent lymphomas are uncurable due to their slow growth. These tumors are more frequent in older patients.
Staging
The staging system used for lymphoid neoplasms is called Ann Arbor staging, which works like this:
- Stage I – cancer in a single region, usually one lymph node and the surrounding area
- Stage II – cancer in more lymph node regions, on one side of the diaphragm
- Stage III – cancer in more lymph node regions, on both sides of the diaphragm
- Stage IV – cancer in one or more extralymphatic organs, like the bone marrow, liver or lung
Also, several modifiers are used:
- E – if there is extranodal involvement
- A – if there are no “B symptoms”
- B – if there are B symptoms
- X – larger than 10 cm mass – called “bulky disease”
Now, let’s get to the specific indolent non-Hodgkin lymphomas.
Chronic lymphocytic leukaemia
Introduction and epidemiology
Chronic lymphocytic leukaemia (CLL) and small lymphocytic lymphoma (SLL) are indolent, mature, non-Hodgkin B-cell neoplasms characterised by accumulation of functionally incompetent lymphocytes. Both CLL and SLL are the same disease with different manifestations. When the disease primarily affects the blood, it’s CLL. When it primarily affects the lymphatic tissue (lymph nodes), it’s SLL.
CLL is the most common adult leukaemia. It mostly affects elderly.
Pathology
The tumor cells of CLL contains high amounts of Bcl-2, however there is no rearrangement of the BCL2 gene involved. Some evidence suggests that the BCL2 gene is upregulated due to loss of regulatory microRNAs.
Two types of CLL exist: the prefollicular type and the postfollicular type. The main difference is whether the immunoglobulin heavy chain gene (IgH) has been mutated or not. The differences are summed up here:
| Prefollicular type | Postfollicular type | |
| IgH gene status | Not mutated | Mutated |
| Has undergone somatic hypermutation | No | Yes |
| Prognosis | Much worse | Better |
| Treatment | More frequently required | Less frequently required |
Source that prefollicular type has not underwent somatic hypermutation and has the worst prognosis: https://www.mayocliniclabs.com/test-catalog/Clinical+and+Interpretive/89008 and http://www.bloodjournal.org/content/120/24/4802?sso-checked=true
Somatic hypermutation is performed by an enzyme called AID. Cancers that have not underwent somatic hypermutation of the IgH gene have worse prognosis because AID will mutate other genes that could make the tumor cells more aggressive instead of mutating the harmless gene IgH. This is not important to know and is just included to explain the above.
A small portion of patients with CLL/SLL will progress into diffuse large B-cell lymphoma (DLBCL); this transformation is called Richter transformation.
CLL/SLL is characterised by CD5+, CD19+, CD20+, CD23+.
Follicular lymphoma
Follicular lymphoma (FL) is an indolent, mature, non-Hodgkin B-cell lymphoma. It’s the most common indolent non-Hodgkin lymphoma and the second most common non-Hodgkin lymphoma overall. It mostly affects older adults, and is characterised by CD19+, CD20+, CD10+, Blc6+, and Blc2+.
It very frequently involves a t(14;18) translocation where the gene BCL2 on the chromosome 18 is fused to the immunoglobulin heavy chain gene (IgH) on chromosome 14. This causes overexpression of the antiapoptotic protein Bcl-2, which contributes to tumor cell survival.
The name “follicular” comes from the fact that the neoplastic cells form “neoplastic follicles” in lymph nodes. The histology of follicular lymphoma can be studied in the slide section.
40% of cases of FL progress into diffuse large B-cell lymphoma.
Mantle cell lymphoma
Mantle cell lymphoma (MCL) is a mature, non-Hodgkin B-cell lymphoma belonging to the “other” group. It originates from mantle zone B-cells and mostly affects adult males. It affects the peripheral blood in only 20% of cases and is therefore not considered a leukaemia. It’s characterised by CD5+, CD19+, CD20+, and cyclin D1+.
It sometimes arises in the gastrointestinal tract, where it manifests as multifocal submucosal nodules that resemble polyps. These can be mistaken for polyps due to other diseases, like familiar adenomatous polyposis, hamartomatous polyps or ulcerative colitis.
MCL involves a t(11;14) translocation where the cyclin D1 gene is fused to the IgH locus. Cyclin D1 is involved in cell proliferation and is overexpressed as a result of this translocation.
It’s an aggressive cancer; the mean survival is 3-5 years.
Marginal zone lymphoma
Marginal zone lymphoma (MZL) is an indolent, mature, non-Hodgkin B cell lymphoma. The name comes from how it develops from marginal zone B-cells. It’s characterised by CD19+, CD20+, and Blc2+.
Three subtypes exist:
- Extranodal marginal zone lymphoma – most common type
- Nodal marginal zone lymphoma
- Splenic marginal zone lymphoma – least common type
Extranodal marginal zone lymphoma, also called MALT lymphoma, arises most commonly from lymphoid tissue in organs such as the stomach, salivary glands, intestines, lungs, orbit or breast. They are associated with chronic infection, such as that seen in H. pylori gastritis, and treating the underlying infection causes the lymphoma to spontaneously resolve. They are also associated with autoimmune disorders, like Sjögren’s (MALT lymphoma in the salivary glands).
Nodal marginal zone lymphoma arises in lymph nodes, most commonly cervical lymph nodes.
Splenic marginal zone lymphoma arises in the spleen or bone marrow.
It’s a bit confusing that you write “lymphoid neoplasms” instead of lymphomas when you’re specifically talking about lymphomas in this topic.
While most lymphoid neoplasms are lymphomas, some, like MGUS and multiple myeloma, are not lymphomas but rather leukaemias.
Change: The table for CLL previously said that the prefollicular type had underwent somatic hypermutation and vice versa. Actually, the opposite is true. It is now fixed, and a source is attached.
🙏🏼😍