Table of Contents
Page created on October 4, 2018. Last updated on January 7, 2022 at 21:45
This topic covers the ways that drugs can affect the body. Most drugs bind to endogenous macromolecules in the body, like receptors, ion channels, enzymes, carrier molecules, structural proteins or DNA. Certain other mechanisms also exist. Let’s go into detail.
Drugs that bind to receptors
Receptors are proteins that transduce a signal when a substance (like adrenalin or acetylcholine) binds to the receptor and mediate some effects to the inside of the cell. Receptors can bind neurotransmitters (like nicotinic receptors), hormones (like insulin receptors) or local hormones (like thromboxane receptors). When we talk about receptors, whatever can bind to a receptor is called a ligand. Neurotransmitters and hormones are ligands for their receptors, and drugs that can bind to a certain receptor are ligands for those receptors as well. Drugs canbind to the receptor instead of the endogenous ligand that usually binds to the receptor in the body.
Drugs that bind to the receptors can have many different effects on the receptors. They can have either agonistic effect, antagonistic effect, inverse agonist effects or act as positive or negative allosteric modulators.
Drugs with agonist effect will evoke the same response as the endogenous compound that the receptor is made for. An example is isoprenaline, a drug which binds to adrenergic receptors just like epinephrine or norepinephrine and evoke a similar response but stronger. Muscarine is another example, which activates muscarinic acetylcholine receptors just like acetylcholine itself does.
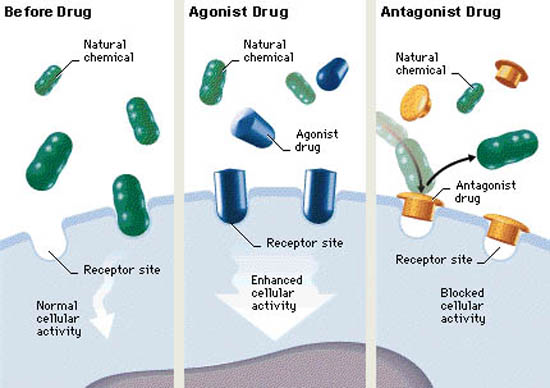
Agonists can be either full agonist or partial agonist. Full agonists have the same response in the tissue as the endogenous ligand, meaning that it’s as “strong” as the physiological molecule that usually binds to the receptor. Partial agonists have “weaker” effects than the endogenous ligand.
Drugs can also have antagonistic effects, which is literally the opposite of agonistic effects. Antagonists bind to the receptor just like agonists, however instead of activating the receptor, the antagonist just sits in the ligand-binding site, doing absolutely nothing. This means that antagonists block agonists from binding to the receptor to evoke their effects, so as an overall effect, antagonists decrease the physiological effect of the endogenous agonist.
All receptors have at least one binding site (for binding the ligand), however some also have other binding sites, where other molecules can bind to. These other binding sites are called allosteric binding sites. Other molecules than the ligand can bind to the allosteric binding site and regulate the activity of the receptor. They can either regulate it negatively, decreasing the effect of the endogenous ligand, or positively, which does the opposite. For example, benzodiazepines bind to an allosteric site on the GABA receptor and increase the effect of GABA on the receptor. In contrast, picrotoxin binds to another allosteric site on the same receptor, but instead decreases the effect of GABA.
We will learn about inverse agonists later.
Drugs that bind to ion channels
Ion channels are membrane proteins that form pores in the cell membrane which allows ion to pass across the membrane. They can be either ligand-gated or voltage-gated. The ligand-gated ion channels are actually receptors, so they belong in the previous category.
Drugs can affect ion channels in two ways. They can either bind to the inside of the channel, blocking the pore so that no ions can pass, or they can bind to regulatory sites on the ion channel itself to either open or narrow the opening to allow more or less ions to flow through.
Local anaesthetics for example block the voltage-gated sodium channels, which stops the pain signals from propagating. Veratridine is an example of a positive modulator of voltage-gated sodium channels, while dihydropyridines are negative modulators of voltage-gated calcium channels.
Drugs that bind to enzymes
To date there are no drugs that bind to enzymes to increase their activity directly. Most drugs that bind to enzymes will inhibit the enzymes. They can do this either reversibly, meaning the drug effect stops after some time, or irreversibly, where the enzymes are permanently destroyed by the drug.
Reversible enzyme inhibitors can be either competitive or non-competitive. Competitive inhibitors bind to the same site as the substrate (they compete for the substrate site) and therefore block the binding of substrate to the enzyme. Non-competitive inhibitors bind to an allosteric site to inhibit the enzyme by making it unable to undergo a conformational change it needs to in order to function properly.
Drugs can irreversibly inhibit enzymes by covalently binding to the enzyme. The irreversible enzyme inhibitors last for much longer than the reversible ones, because the body must synthesize new enzyme molecules to replace the ones that are irreversibly inhibited by the drug. Some poisons called organophosphates irreversibly inhibit acetylcholinesterase, for example.
Drugs can inhibit enzymes in one other way. Some drugs resemble a metabolite normally found in the body so that the enzyme will use the drug as a substrate instead of the originally intended metabolite. This interferes with the physiological pathway of the enzyme. Take the enzyme DOPA decarboxylase for example. Usually it converts DOPA to dopamine, but the drug methyldopa will act as the substrate instead and therefore be converted to methyldopamine which is later converted to methylnoradrenaline. This means that methyldopa “competes” with DOPA for the enzymatic activity of DOPA decarboxylase. This means that the enzyme is less available for the natural substrate, which essentially inhibits the enzyme. After the drug has been modified by the enzyme, it will become a different compound that may have other effects on the body, a so-called false metabolite. Methylnoradrenaline is such an example.
While there are no drugs that can stimulate enzymes directly, there are some that stimulate them indirectly. We can’t stimulate guanylyl cyclase directly, but the drug nitroglycerine can be used to stimulate it indirectly. Nitroglycerine will give off NO which will stimulate the guanylyl cyclase enzyme.
Drugs that bind to transporters
Transport molecules are proteins that transport molecules across a lipid membrane. They can be inhibited reversibly, irreversibly or by a false substrate.
A type of drug called cardiac glycosides reversibly inhibit the Na+-K+ ATPase so that the heart will contract harder.
A more known type of drug called proton pump inhibitors irreversibly inhibit the H+-K+ ATPase in the parietal cells in the stomach, causing the stomach acid to have a higher pH. The drug will covalently bind to the ATPase, which permanently inhibits it.
The third type of transporter-binding drugs are actually substrates for the transporter. Like with the enzymes, this type of drug will use the transporter protein to get transported, which makes the transporter less available for its natural ligand.
Drugs that bind to other types of proteins and DNA
Drugs can also bind to structural proteins or immunological proteins to mediate other effects.
Certain anticancer drugs bind to and alkylate DNA.
Drug mechanisms that don’t bind to proteins or DNA
Drugs called antacids neutralize the stomach acid simply by being bases themselves, which neutralize the acid by simple acid-base interaction.
Drugs can work via osmosis as well. Osmotic laxatives aren’t even absorbed by the body, but simply their presence in the GI tract will alter the osmotic balance, causing fluid to remain in the GI tract instead of being absorbed. Osmotic diuretics alter the osmotic balance in the kidney.
Great work however, the article should be referenced for easy citation and research.
Thank you. Having the works cited would be ideal but it’s not something I would have the time to do.
Hello,
I just wanna make sure is the proton pump inhibitor works on the H+/K+ ATPase of chief cells or parietal cells of the stomach ?
Thank you for your effort.
Sorry, I always mix up the two. It’s fixed now.
Hei bro
One question. You say “to date there are no drugs that bind to enzymes to increase their activity directly.” But don’t CYP inducers do this?
Thanks!
CYP inducers increase the genetic expression of the CYP enzymes. They don’t directly bind to them and increase their activity.
Got it! Thank you again