Table of Contents
Page created on September 26, 2018. Last updated on January 7, 2022 at 21:46
Summary
- Occupancy shows the fraction of the total number of receptors in the tissue that is occupied by the drug. It’s determined by the constant p.
- A drug’s affinity to its receptor is determined by the constant Kd. When Kd is low is the affinity high. A high affinity is always good and has no drawbacks.
- Efficacy is a measure of how “effective” the drug is. It measures how large of a cellular response the drug can induce in the tissue, which roughly translates to how severe of a condition or symptom the drug can treat. The drug’s intrinsic activity determines its efficacy.
- Intrinsic activity (IA) is a measure of the efficacy of a drug compared to the most efficacious drug with the same function. IA ranges between 0 and 1.
- Agonists have an intrinsic activity of 1
- Partial agonists have an intrinsic activity of more than 0 but less than 1
- Antagonists have an intrinsic activity of 0
- Potency is a measure of how much of a drug you need to get the desired effect. It’s measured by the constant ED50. The lower the ED50, the more potent the drug is. It depends on the affinity and the intrinsic efficacy.
- A high potency is always good because when a drug is potent even a low concentration can give a satisfactory therapeutic effect. By allowing the use of low concentrations potent drugs reduce the risk for adverse effects.
- Receptor reserve is a measure of how many receptors in the tissue that are not occupied by the drug even when the drug has reached its maximum effect.
- Intrinsic efficacy is a measure of the receptor-activating ability of an agonist. It is measured by the constant ε, and it depends on the intrinsic activity and receptor reserve. A higher intrinsic efficacy is always good.
This topic is theory-heavy so hold on tight.
In this topic we study the agonist-receptor interaction, that is, what determines how an agonist affects the receptor and how can we differentiate different agonists.
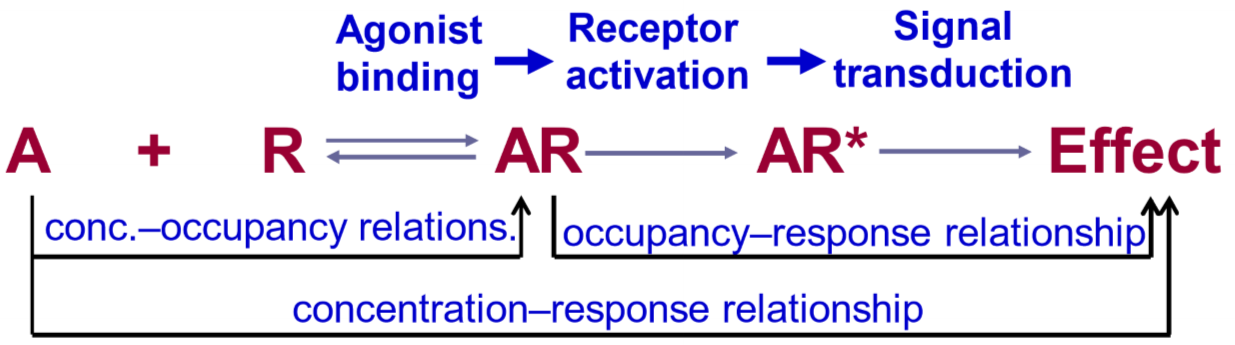
This figure is scary. The A is the agonist, while the R is the receptor. In order for anything to happen, the agonist must bind to the receptor to form AR. This binding is not permanent, meaning that there is an equilibrium between the number of free agonists (A) and receptors (R) and bound agonist-receptors (AR), which is why the arrow goes both ways. After A has bound R, the receptor will become activated (AR*). When the receptor is activated, it will cause an effect.
As we can see on the figure, the relationship between the agonist concentration and how many agonists are actually bound to a receptor (AR) is called the concentration-occupancy relationship. The relationship between how agonists bound to the receptor cause their effect is called the occupancy-response relationship. The sum of these two relationships, the relationship between the agonist concentration and the resulting tissue effect is called the concentration-response relationship. We’ll discuss the three separately.
Concentration-occupancy relationship
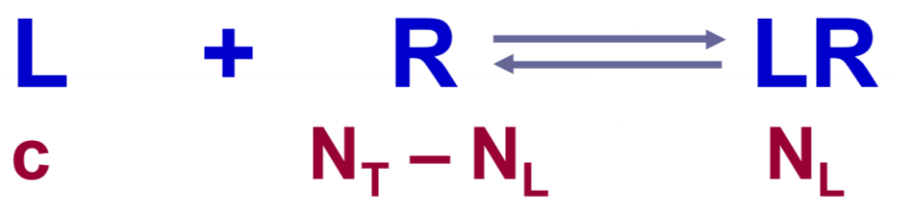
This is the same equation as in the previous figure, however we use ligand instead of agonist in this case because this relationship doesn’t only count for agonists, but for antagonists as well. For our use case we can think of the ligand as a drug.
Now, we’re interested in knowing the answer to the following question: How does the concentration of the drug affect how many receptors are actually bound by the drug? We want to know this because it turns out that when we apply drugs to tissues, the drugs don’t occupy all the receptors. Then, what determines how many drug molecules will bind to the receptors?
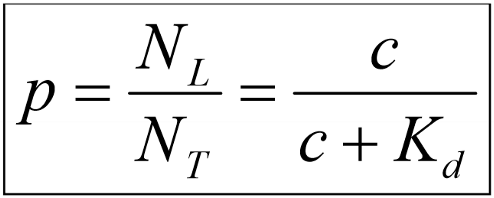
We’re interested in finding out what parameters determines how big the fraction of receptors that have bound a ligand is. Like, what determines if only 50% of the receptors are occupied by a ligand, or 75%, or 100%? The fraction (p) of occupied receptors can be calculated by calculating NL/NT. We call this p for occupancy. Mathematical magic concludes that this fraction (p) is equal to c/(c+Kd). Kd is a constant that is characteristic for a single drug. Each drug has its own Kd. The Hill-Langmuir equation therefore tells us that the fraction of occupied receptors (p) is determined only by the concentration of the drug and a drug constant. Makes sense, right?
Mathematics show that the constant Kd is the concentration that the drug must be given in in order to occupy exactly 50% of the available receptors. That means that if a drug has a Kd of 0.24M, then this drug must be given in a concentration of 0.24M in order to occupy half of the receptors in the given tissue.
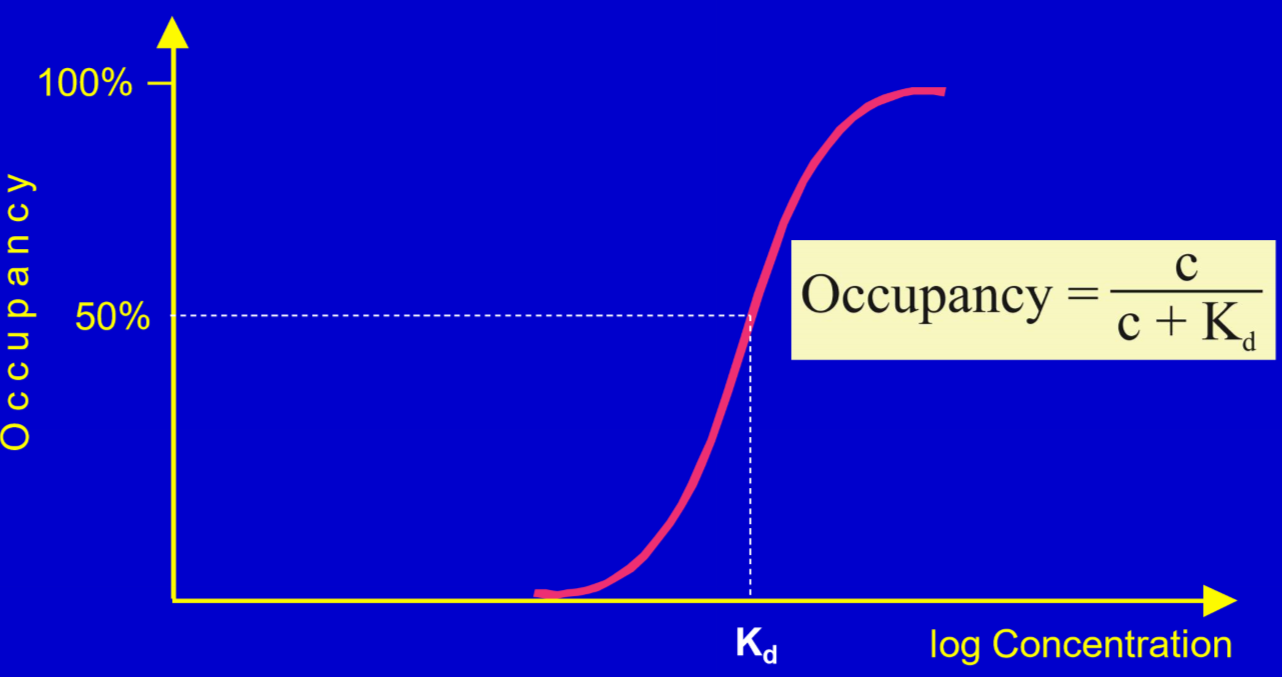
From the graph, we can see that any drug can occupy 100% of the receptors as long as the concentration is high enough. Likewise, when the drug concentration sinks, the occupancy sinks along this sigmoidal curve.
Kd characterizes the drugs tendency to bind the receptor, also called the affinity. The lower the Kd, the higher the affinity, meaning that they are inversely correlated. A high affinity is always good, because when the drug has a high affinity then a lower concentration of the drug is needed for it to occupy the desired fraction of receptors.
Concentration-response relationship
It’s easy to determine the concentration-response relationship. In humans, we do it like this:
Let’s say that we want to determine the concentration-response relationship of a new vasodilator. We determine a satisfactory clinical effect, like “the blood pressure of the patient decreased by 10 mmHg or more”, and then give the drug to many patients. First, we give it at a very low concentration. At a low concentration, the drug is unlikely to give the desired clinical effect in many of the patients, however we write it down if some of them do. Then, we increase the dose slightly, then do it again and repeat. As the dose increases, the fraction of patients that reach the desired effect will increase, until at one point, the drug concentration is so high that every single patient have reduced their blood pressure by 10 mmHg.
Then, we plot the data into a graph. It’s going to look like this:
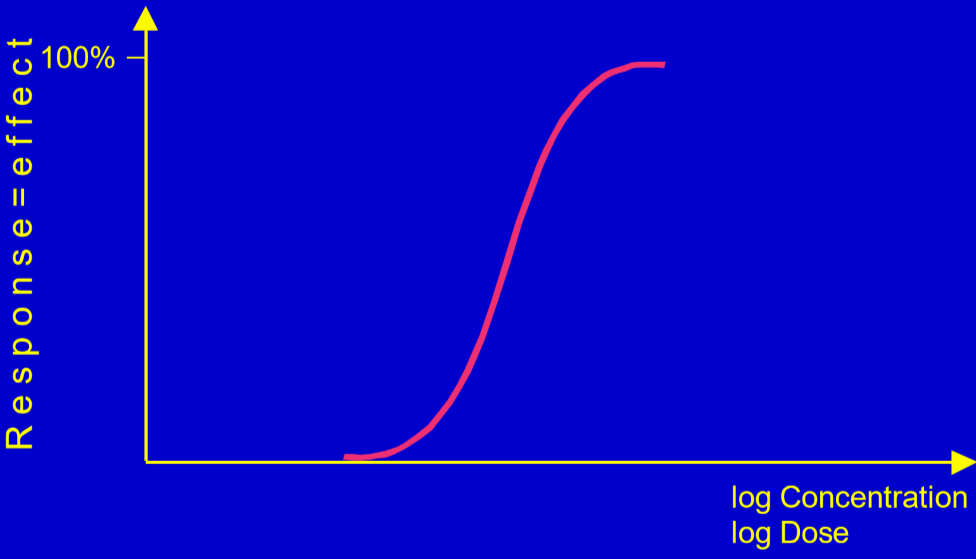
We’re going to introduce three new terms: efficacy, intrinsic activity and potency.
Efficacy (pronounced effikacy) can be though of as effectiveness. It shows how big the effect of the drug has the potential to be, if the concentration is high enough. For an antihypertensive drug the efficacy of the drug would tell us how much the drug can reduce the blood pressure by.
The efficacy is determined by the drug’s intrinsic activity (IA). IA of a drug ranges between 0 and 1 and shows how big the effect of the drug is, compared to the largest effect achievable in the tissue by the most efficacious drug. Efficacy determines how severe symptoms can be treated with the drug. Let’s look at an example.
We have a group of patients with cancer-related pain, which is very severe. We divide the group into two and give one group paracetamol for the pain and the other group morphine. It’s pretty certain that no matter the dose of paracetamol it will never be enough to treat the pain the patients have, however the morphine probably will. This is because paracetamol has a lower efficacy than morphine in this case; paracetamol just isn’t efficacous enough as a painkiller to treat such severe pains.
Potency is the measure of how large of a dose of a drug is needed to achieve the desired effect. It’s determined by a constant called ED50 (or EC50, they mean the same in this context). This constant determines the concentration of the drug needed to achieve 50% of the maximum possible effect of the drug. This means that more potent drugs need a lower concentration to reach the same effect as a similar but less potent drug. When ED50 is low, the drug is potent.
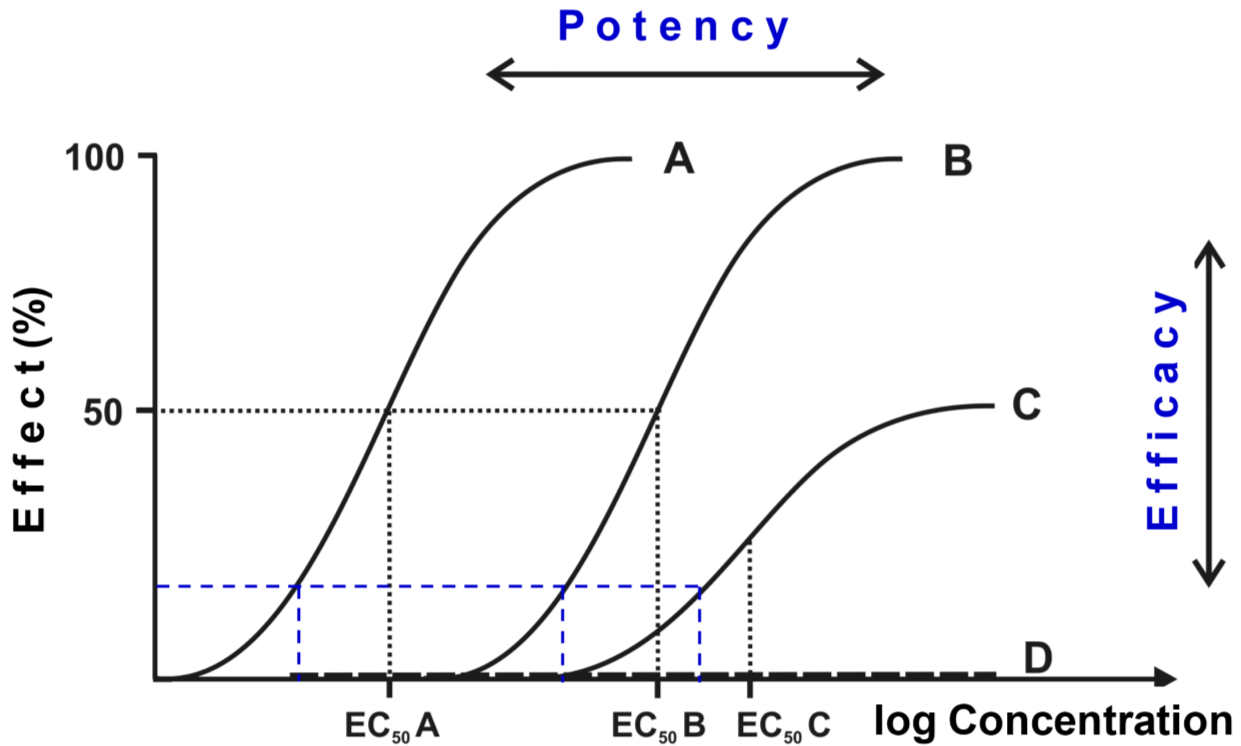
In the graph above the curves of four different drugs can be seen. The figure shows that the curves’ position along the x-axis shows their potency, while their height shows their efficacy. Drug A is the most potent, followed by drug B and lastly drug C. Drug A and B have the same efficacy, while drug C has a lower efficacy. Drug D has no efficacy (it’s probably an antagonist).
In this example, drug A and B are full agonists, because drug A and B can reach 100% of the achievable effect in the tissue, while drug C can only reach about 50%, meaning it is a partial agonist. Drug A and B will therefore have intrinsic activity = 1 (= 100%), while drug C has IA = 0.5 (= 50%). All drugs that have a lower than 100% efficacy are partial agonists, meaning they can’t give the maximum possible effect of the tissue. Drug D have an IA of 0.
A high potency is always a good thing, because when a drug is very potent it can be given in a low dose, which can reduce non-receptor mediated side effects. Efficacy is a different story however, as it depends on what we need. Think back to the painkiller example: we wouldn’t use morphine to treat a headache!
The last thing we must talk about in this case is the margin of safety. Looking back to the concentration-response curves we need to take a look at their slopes, that is, how steep the curve is around the middle. When a curve is very steep, it means that a small increase in dose will give a larger effect, while if the curve is gentle (not steep), then a small increase in dose won’t give a much larger effect. This means that drugs with gentler slopes are safer, as the drug dosage can be varied somewhat without a large change in effect.
Occupancy-response relationship
Recall that the Kd constant shows the drug concentration where half the receptors are occupied by the drug, and that the ED50 constant shows the drug concentration where half of the possible effect is reached. Intuition may say that they should be equal, but they’re not. ED50 is always smaller than (or equal to) Kd, meaning that the drug concentration needed to give 50% effect is lower than the concentration needed to saturate 50% of the receptors. The same also goes for 100%: the drug concentration needed to give 100% effect is lower than the concentration needed to occupy 100% of the receptors. What does this mean?
It means that when we give a drug in the concentration that gives 100% effect, there are many receptors that are not occupied by the drug! We introduce a term called receptor reserve, defined as the fraction of receptors that are apparently not needed for the maximal effect. Receptor reserve can be found by plotting a drug’s effect against its occupancy, like this:
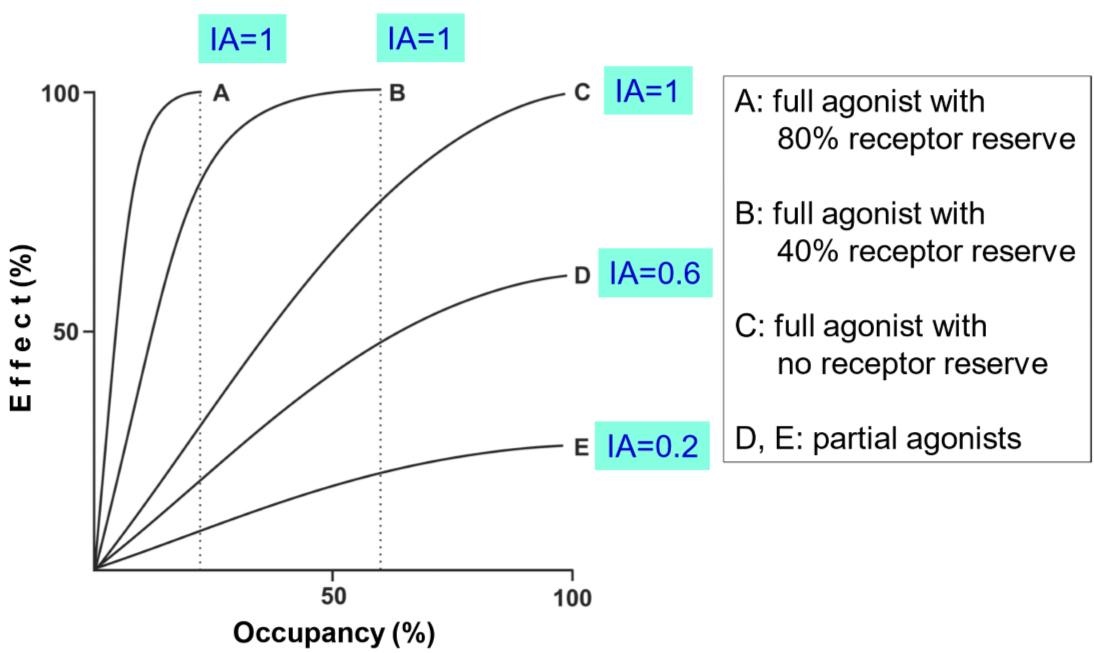
Here we have plotted the occupancy-response relationship of five different drugs. From this graph, we can determine the drugs’ efficacy (their intrinsic activity, IA) and their receptor reserve. The IA is where the highest point of the graph is on the y-axis. To find the receptor reserve, we need to see at what occupancy the drug can achieve 100% effect. Drug A reaches 100% effect at around 20% occupancy. That means that 100 – 20 = 80% of the receptors are available, meaning that for drug A, the receptor reserve is 80%. Drug B achieves 100% with around 60% occupancy, meaning that it has a receptor reserve of around 40%. C achieves 100% effect with 100% occupancy, meaning that it has no receptor reserve. D and E never even reach 100% effect, so they have no receptor reserve. We can see that their efficacy is below 100%, so they’re both partial agonists.
There are two mechanisms that can explain the how a maximum effect can be reached without all receptors being occupied:
- High signal amplification in the receptor, so even if not all receptors are activated the response will still be 100%
- High receptor density, meaning that there are so many receptors that some are left unbound after 100% effect has been reached
We introduce yet another term: intrinsic efficacy (not to be confused with intrinsic activity). Intrinsic efficacy is a measure of the receptor-activating ability of an agonist. It describes how efficiently receptor occupancy translates into a biological effect or change.
Intrinsic efficacy depends on the drug’s receptor reserve and intrinsic activity. The symbol of intrinsic efficacy is ε (epsilon). On occupancy-response relationship graphs, the intrinsic efficacy increases upwards and to the left. On the previous graph, the intrinsic efficacy of the five drugs are different. A has the highest, then B, C, D and E has the lowest.
A higher intrinsic efficacy is a good thing, because drugs with higher intrinsic efficacy have higher receptor reserves and higher intrinsic activity. Intrinsic efficacy can be used to differentiate the usefulness of different full agonists.
You wrote “Potency is a measure of how much of a drug you need to get the desired effect. It’s measured by the constant ED50. The lower the ED50, the more potent the drug is. It depends on the affinity and the intrinsic efficacy”
Instead of ED50 – do you mean constant EC50?
If you see one of the other comments, I’ve already explained how they’re interchangeable in this topic.
Why is receptor reserve important pharmacologically? (how is it important to patients? what can it be used for?)
That question applies to much of the seminar knowledge from pharma 1. From what I know, it’s not at all important to know clinically, but they ask it in the exam.
This definition of ED50 is wrong. The ED50 is the concentration needed to elicit the wanted effect in 50 % of the population.
It’s not wrong; it’s just not the only definition of ED50. You’re right, ED50 can also mean what you said. But in the context of pharmacodynamics, it means what I wrote. See the seminars, the book (chapter 2), here or here.
Hi! Does the intrinsic activity or intrinsic efficacy determine the efficacy? in the summary you have written intrinsic efficacy, but in further down in the text intrinsic activity.
Fixed now
This is Bojack… Horseman.. obviously
The memelord himself
Thank you for taking the time to write notes that are enough for exams.
This has been very helpful and selfless of you.
Thank you for helping me .
Glad I could help <3
«EC50 is always smaller than (or equal to) Kd, meaning that the drug concentration needed to give 50% effect is lower than the concentration»
Do you mean ED50 here? 🙂
They’re interchangeable in this context, but I changed it now for consistency.
Commendable effort is recognised for these notes; however I must emphasise that the layout on this topic is very misleading. Please revise this topic and re-write to demonstrate the various pharmacological jargon to the lay man who would be misinformed otherwise.
I agree that the layout isn’t optimal and I’m sure you’ll find more topics that have the same problem. The layout somewhat follows the layout of the lecture or seminars they’re based on.
I will not rewrite it as I’m not sure I could do it better if I tried and I don’t want to use valuable time on that. I have already simplified it compared to the original seminar and in my opinion have I not left out anything that would be important. The topic isn’t written for laymen anyway.
If you have any more specific points you would want improved then could I take a look.