Table of Contents
Page created on January 5, 2019. Last updated on January 7, 2022 at 21:48
Summary:
- The following factors influence GI tract absorption: GI motility, gastric emptying, luminal pH, the presence of food together with the drug
- Drugs which are weak acids (aspirin) are absorbed in the stomach
- Drugs which are weak bases (amphetamine) are absorbed in small intestine
- Absorption of lipophilic drugs increases with a fat-rich meal
- Hydrophilic drugs must be absorbed with fascilitated diffusion
- Calcium intake impairs absorption of tetracyclines, fluoroquinolones
- Oral bioavailability is a fraction of how much of the drug that reached the systemic circulation and how much of the drug that was given orally
- The main factor which reduces oral bioavailability is first pass metabolism, but also poor absorption and degradation of the drug in the GI tract
- Presystemic elimination refers to any elimination of the drug which occurs between taking the drug orally and the drug reaching the systemic circulation
- It’s mostly due to first pass metabolism, metabolism of the drug in the intestinal mucosa and liver
- It can also be due to efflux proteins which efflux the drug back into the GI lumen
- A high first pass metabolism is always a bad thing, except when the drug acts on the liver (like statins)
- The oral, nasal and rectal mucosa don’t have transport proteins for drugs. All drugs applied here must be absorbed by diffusion (must be lipophilic)
- Drugs which are peptides (like pituitary hormones) are often given nasally, to prevent breakdown by peptidases in the intestines
- The skin don’t have transport proteins either. All drugs to be absorbed through the skin must be lipophilic.
- Absorption through the lungs is good because:
- The lung has a huge absorptive surface area
- The distance from the alveoli to the capillaries is very short
- The lungs have very large blood flow
- This is mostly only important for general anaesthetics, the only important drugs which are given by inhalation and which should enter the systemic circulation
- Most drugs given by inhalation are meant to act locally in the lungs and airways, like bronchodilators and anti-inflammatory drugs
Absorption from the GI tract
In topic 8 we discussed transport across membranes, and that’s obviously important for absorption as well. Lipophilic drugs are absorbed by free diffusion while hydrophilic drugs are absorbed by fascilitated diffusion, often with the help of a transport protein.
What influences the rate of absorption from the GI tract? Four factors do:
- The motility of the GI tract, including the gastric emptying and intestinal propulsion
- The luminal pH
- The quantity of the luminal content, i.e. how much partially digested food is present together with the drug
- The quality of the luminal content, i.e. what type of partially digested food is present
GI motility is the first factor. There are two mechanisms at work here: the gastric emptying, which is how long it takes for the stomach to release content into the duodenum, and intestinal propulsion, which determines how long the drug will stay in the intestines
When gastric emptying is delayed is the drug absorption also delayed (except in the cases where the drug is absorbed mainly from the stomach of course). Fatty meals, certain drugs like anticholinergics and some diseases causes delayed gastric emptying and therefore delayed intestinal absorption.
Gastric emptying is fastened when in a fasting state, when there is a large intake of water and in certain drugs. These factors fasten the intestinal absorption.
Decreased intestinal propulsion means more time to absorb drugs. The seminar doesn’t have any examples for why it would be decreased so 🤷♂️. I guess ileus?
Increased intestinal propulsion means less time to absorb drugs. This is typical in diarrhoea.
Luminal pH is the second factor and is important for the reasons we discussed in topic 8. As a summary: drugs that are weak organic acids (like aspirin) are mainly absorbed in the stomach and only partially in the intestine, because when acids are in a low pH solution are most of the acid molecules in their uncharged form (HA).
Drugs that are weak organic bases (like amphetamine) are not absorbed in the stomach, only in the small intestine. When bases are in a basic solution are most of the base molecules in their uncharged form (B).
Quantity of luminal content is the third factor. Some drugs prefer to be taken while fasting, like erythromycin. Other drugs benefit from the increased splanchnic blood flow that comes with eating food, like propranolol.
The quality of the luminal content is the last factor. It’s important to know that only dissolved drug molecules can be absorbed – solid particles cannot. It’s therefore important to give the drug an environment that it can dissolve itself in. What that type of food is depends on the drug itself. Here are some examples:
- Certain drugs, like itraconazole, must be dissolved in acid. In cases where the acidity of the stomach is low must this drug be taken with an acidic beverage.
- Lipid-soluble drugs prefer to be taken with a fatty meal.
- Fatty meals slow gastric emptying and may decrease absorption of certain drugs
- High protein meals can interfere with the absorption of certain drugs, if these drugs are absorbed by enterocytes through the same transporters as amino acids and peptides. For example, L-DOPA is absorbed by the transporter LAT-1, which also transports amino acids.
- High intake of fruit juices can be problematic because molecules in the juices can inhibit certain transporters
- High intake of Ca2+, like with milk or yoghurt, can be problematic because certain drugs create complexes with Ca2+ which makes them impossible to absorb. This includes tetracyclines and fluoroquinolones.
In some cases do we want to decrease GI absorption. For example, in hypercholesterolaemia we want to inhibit bile acid absorption, in hyperkalaemia we want to inhibit K+ absorption and in renal failure we want to inhibit phosphate absorption. Compounds for these uses are available.
Charcoal is used to decrease the absorption of drugs after an oral overdose. Charcoal binds to the drugs and decreases their absorption and increases their elimination.
Oral bioavailability
The oral bioavailability is a measure of how much of an orally taken drug that reaches the systemic circulation in unchanged form. To be more precise is it a fraction, and the symbol we use for it is F. It’s a fraction between how much of the drug that reached the systemic circulation and how much of the drug that was given orally.
The oral bioavailability is a number that is characteristic for each drug. If a drug has an F = 1 does that mean that 100% of the oral dose reached the systemic circulation unchanged. If it has F = 0.5 does that mean that only 50% of the oral dose did.
Why don’t all drugs have 100% oral bioavailability? Multiple factors play a role:
- It’s possible that not all the drug molecules are liberated from the tablet or capsule for some reason. This is a result of poor drug formulation.
- Degradation of the drug in the lumen by digestive enzymes or acid
- Poor solubility of the drug in intestinal fluids
- Poor diffusibility of the drug, because the drug is very hydrophilic
- First pass elimination, or pre-systemic elimination of the drug
1 and 2 warrant no further explanation, and 3 and 4 have already been discussed.
First pass metabolism:
When drugs are taken orally must they pass through two metabolically important areas before they reach the systemic circulation: the intestinal mucosa and the liver. Both organs have CYP450 enzymes and other enzymes that can inactivate the drug before it reaches systemic circulation. The most abundant CYP450 enzyme in the enterocytes is CYP3A4 which inactivates many drugs like cyclosporin and midazolam. These drugs therefore have F = 0.3 and F = 0.4, respectively.
First pass metabolism doesn’t just include enzymatic inactivation of the drug. Enterocytes may efflux the drug molecules back into the GI lumen, often by the use of Pgp/MDR1.
The F value for a drug is calculated by giving healthy volunteers the drug both IV and orally at different times and comparing the concentration of the drug over time in both cases. That gives a graph that looks like this:
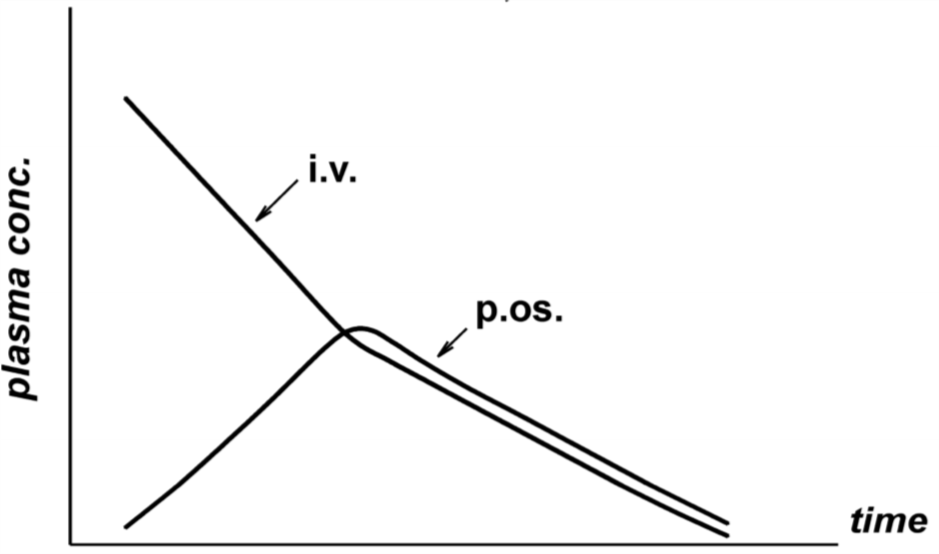
p.os stands for per os, which means “orally”. Now, to determine the F value must we divide the area under the curve for the per os graph with the area under the curve for the IV graph. F = AUCp.os : AUCi.v. In other words, we must divide the area marked blue with the area marked with red.
I don’t think this is very important to know.
Most drugs are given with the intention that it should reach the systemic circulation in as high amount as possible. So, a low F value is unfavourable for most drugs. However, some drugs target the liver and GI tract, so for these drugs is it good that they don’t reach the systemic circulation completely. HMG-CoA reductase inhibitors (statins) for example act on the liver, so for them is it good to be taken up into the liver.
It’s possible to increase the oral bioavailability of a drug by administering other drugs simultaneously that inhibit whatever protein that is the cause of the low bioavailability. For example, itraconazole is a CYP3A4 inhibitor, so if you administer it with drugs that are metabolised by CYP3A4, like cyclosporin, will less cyclosporin be metabolised and so more of it will reach the systemic circulation.
Pgp inhibitors like ritonavir may also be used, to increase bioavailability of drugs which are substrates for Pgp, like protease inhibitors.
Absorption from the oral, nasal and rectal mucosa
Unlike the rest of the GI tract the oral and rectal mucosa were never intended to absorb stuff. The same goes for the nose. These organs therefore don’t have any transporter molecules to absorb drugs – all drugs must be absorbed by diffusion. Therefore must all drugs that are to be absorbed through these mucosas be lipophilic.
There are two major advantages in delivering drugs this way:
- The drug is not subjected to stomach acid and digestive enzymes
- The drug avoids any kind of first pass metabolism
Many peptide-based drugs are given this way, because they would be broken down by peptidases in the GI tract.
Giving drugs as a rectal suppository is an advantage in several cases:
- When oral administration is difficult, like for babies or vomiting patients
- When the GI tract mucosa is vulnerable, like due to ulcers
- When the drug has significant first pass metabolism if taken orally
Absorption through the skin
Certain drugs can be given through the skin. This applies especially to local anaesthetics, but other drugs can be applied as well, like ointments, powders or transdermal patches.
For drugs to diffuse past the epidermis must they again be lipophilic. If the drugs are lipophilic enough and potent enough can they be administered as patches that give systemic effects. This can be done with nitro-glycerine, nicotine and oestradiol for example.
These patches must have a large concentration of the drug to create a large concentration gradient to serve as a driving force for the drug to diffuse. If the concentration isn’t large enough the concentration gradient will be small, which reduces absorption. The area of the topical patch is also significant – the bigger the patch, the higher the dose rate, the rate of which the drug is given over time.
The thickness of the stratum corneum is significant in this absorption. The thicker the layer, the lower the absorption. S. corneum is the thickest at the sole and palm, and the thinnest at the forehead, submandibular area and the scrotum.
Infants don’t yet have stratum corneum, so they’re more predisposed to chemical absorption through skin.
The dermal circulation also plays a role. It is increased if the skin is rubbed, in inflammation and in warm environments. This promotes absorption.
Absorption from the lung
Sometimes it’s good to give things by inhalation, like:
- Gases like O2, N2O, CO2
- General anaesthetics
- Vasodilators
However, absorption through the lung is bad in some cases:
- Gases like CO, HCN, H2S
- Organic solvents like chloroform
Absorption through the lungs is good because:
- The lung has a huge absorptive surface area
- The distance from the alveoli to the capillaries is very short
- The lungs have very large blood flow
These factors make pulmonary absorption suitable for many things, both for systemic use and for local use.
If drugs are given by inhalation with the intention of reaching the systemic circulation should the drug be as absorbable as possible, i.e. as lipophilic as possible. Examples of this are general anaesthetics and amyl nitrate for angina.
If the drugs are given with the intention of treating a condition of the bronchial mucosa or smooth muscle, such as asthma, cystic fibrosis or COPD, is absorption undesirable because we want the drug to stay in the alveoli. Examples of this are bronchodilators and glucocorticoids for asthma, and Na+ channel antagonists for cystic fibrosis.
is there a topic for Pharma myocardial ischemia or nitrates?
Topic 25 “antianginal drugs” may cover what you’re looking for.